http://www.chemistrymag.org/cji/2005/076042ne.htm |
Jun.1, 2005 Vol.7 No.6 P.42 Copyright |
Direct syntheses of novel calix[4]thiocrown and calix[6]thiocrown
Yang Fafu,
Guo Hongyu, Lin Jianrong
(College of Chemistry and Materials Science, Fujian Normal University, Fuzhou 350007,
China)
Received on Apr.3, 2005; Supported by the National Natural Science Foundation of China (No. 20402002) and Fujian Natural Science Foundation of China (No. E0220002)
Abstract By reacting calix[4]arene or
calix[6]arene with diethylene thio-glycol bischloroacetates, a new 1, 3-bridged
calix[4]thiocrown 3 and the first example of calix[6]thiocrown 4 were
directly synthesized in moderate yields.
Keywords calixcrown, thio-, synthesis
1 INTUODUCTION
Calixcrowns are playing an important role among all kinds of calixarene derivatives
due to their superior recognition capabilities and complexation selectivities than that of
calixarenes and crown ethers [1, 2]. The complexation capabilities can be tuned
by introducing some functional groups into crown ether segment, for example,
calix-thio-crowns show outstanding recognition selectivity towards soft cations[3].
Up to now, several calix[4]thiocrown were synthesized by indirect method but the total
yields were low[3, 4], and no successful method to synthesize calix[6]thiocrown
was reported. For promoting the research of calix-thio-crowns, it was necessary to find a
facile method to synthesize calix-thio-crowns. In this paper, we wish to report a direct
method to synthesize two novel calix-thio-crowns: the first example of calix[6]thiocrown
and a new calix[4]thiocrown.
2 RESULTS AND DISCUSSION
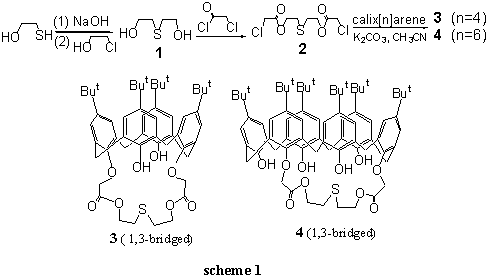
The synthetic route was shown in Scheme 1. Diethylene thio-glycol bischloroacetates 2
was synthesized by reacting compound 1 with chloracetyl chloride. Treating
compound 2 with calix[4]arene in the presence of K2CO3 as
base and KI as catalyst in acetonitrile, 1,3-bridging calix[4]thiocrown containing ester
groups 3 was isolated in yield of 42%. Using calix[6]arene instead of calix[4]arene
in this reaction, no 1,2- or 1,4-bridging calix[6]arene but only 1,3-bridging
calix[6]thiocrown 4 was isolated in yield of 28%. Using other solvents such as
benzene, DMF or tetrahydrofuran instead of acetonitrile, the calix[6]arene did not take
part in reaction or the reaction were complicated very much and no products were separated
successfully. Comparing with indirect method of synthesizing calix[4]thiocrowns, this
direct method was not only simple but also the yields were moderate among all kinds of
calixcrowns. Moreover, differing from indirect method which was not suitable for
synthesizing calix[6]thiocrown[5], this direct method was used successfully in
the syntheses of calix[6]thiocrown.
The structures of compounds 3 and 4 were characterized by
FAB-MS spectra, elemental analyses, 1H NMR spectra. The 1H NMR
spectrum of 3 showed two singlets (1:1) for the tert-butyl groups and two
pairs of doublets (1:1) for methylene bridge protons, which certainly indicated that
compound 3 was 1,3-bridged and adopted cone conformation. In the 1HNMR
spectrum of 4, four singlets (2:2:1:1) for the tert-butyl groups and six
singlets (1:1:1:1:1:1) for the calixarene aromatic protons indicated that calix[6]arene
moiety was intramolecularly bridged at 1,3-position[5]. Due to the overlapped
signal for ethylene protons of crown ether chain and methylene bridge protons of
calix[6]arene, it was difficult to confirm the conformation of compound 4. The
complexation capabilities of novel calix-thio-crowns are under investigation currently.
Melting points were uncorrected. 1H NMR spectra were recorded in CDCl3 on a Bruker-ARX 300 instrument at room temperature. Using TMS as an internal standard. FAB-MS spectra were obtained from a Kratos MS80RF mass spectrometer. Elemental analyses were performed at Vario EL III Elemental Analyzer. All solvents were purified by standard procedures. p-tert-butylcalix[4, 6]arene were prepared according to the published procedures.
3.1 Synthesis of diethylene thio-glycol bischloroacetates 2
Chloracetyl chloride (0.2mol) was dropped in an ice bath flask of diethylene thio-glycol 1 (0.1 mol) under stirring and the mixture was stirred at 70ºC for half an hour after dropping over. Then 20mL water and 50mL CHCl3 was added and the organic layer was separated, washed by distilled water for three times, dried by MgSO4, and filtered. Compound 2 was obtained as straw color oil in yield of 91% by distilling solvent under reduced pressure. 2: 1HNMR (300 MHz, CDCl3): 2.83(t, J = 7.2Hz, 4 H, SCH2), 3.98(t, J = 7.2 Hz, 4 H, OCH2), 4.23(s, 4 H, OCH2CO). MS(FAB): 275 (M+, 100%), Anal. calcd for C8H12O4Cl2S: C, 34.91; H, 4.36%. Found: C, 34.86; H, 4.41%.
3.2 Synthesis of p-tert-butylcalix[4]thiocrown containing ester groups 3
A mixture of p-tert-butylcalix[4]arene (1mmol), diethylene thio-glycol bischloroacetates 2 (1.1mmol), K2CO3 (3 mmol), KI (2.2mmol), was stirred in refluxing acetonitrile (100ml) for two days under N2. After distilling off the solvent, the residue was treated with HCl (10%, W/V) and extracted with CHCl3. The organic layer was separated, dried (MgSO4), and then filtered, concentrated. By column chromatography on silica gel (100-200 mesh, 90% CH2Cl2, 10% Et2O), compound 3 was obtained in yield of 42%. 3: m.p. 217-220oC 1HNMR (300 MHz, CDCl3): 1.02(s, 18 H, C(CH3)3), 1.21(s, 18 H, C(CH3)3), 2.80(t, J=7.2Hz, 4 H, SCH2), 3.38(d, J=13.6Hz, 4 H, ArCH2Ar), 3.97(t, J = 7.2 Hz, 4 H, OCH2), 4.22(s, 4 H, OCH2CO), 4.27(d, J=13.6Hz, 4 H, ArCH2Ar), 6.65(s, 4 H, ArH), 7.01(s, 4 H, ArH), 7.19(s, 2 H, ArOH). MS(FAB): 850(M+, 60%), Anal. calcd for C52H66O8S: C, 73.41; H, 7.76%. Found: C, 73.35; H, 7.79%.
3.3 Synthesis of p-tert-butylcalix[6]thiocrown containing ester groups 4
A mixture of p-tert-butylcalix[6]arene (1mmol), diethylene thio-glycol bischloroacetates 2 (1.1mmol), K2CO3 (3 mmol),KI (2.2mmol), was stirred in refluxing acetonitrile (100 mL) for two days under N2 atmosphere. After distilling off the solvent, the residue was treated with HCl (10%, W/V) and extracted with CHCl3. The organic layer was separated, dried (MgSO4), and then filtered, concentrated. By column chromatography on silica gel (100-200 mesh, 85% CH2Cl2, 15% Et2O), compound 4 was obtained in yield of 28%. 4: m.p. 242-244oC 1HNMR (300 MHz, CDCl3): 1.20(s, 9 H, C(CH3)3), 1.22(s, 9 H, C(CH3)3), 1.25(s, 18 H, C(CH3)3), 1.27(s, 18 H, C(CH3)3), 2.80(t, J=7.2Hz, 4 H, SCH2), 3.36(d, J=13.9Hz, 2 H, ArCH2Ar), 3.42(d, J=14.2Hz, 2 H, ArCH2Ar), 3.87-3.98(m, 6 H, ArCH2Ar and OCH2), 4.18-4.34(m, 6 H, OCH2CO and ArCH2Ar), 4.50(d, J=13.9Hz, 2 H, ArCH2Ar), 4.64(d, J=14.2Hz, 2 H, ArCH2Ar), 6.97(s, 2 H, ArH), 7.03(s, 2 H, ArH), 7.05(s, 2 H, ArH), 7.09(s, 2 H, ArH), 7.11(s, 2 H, ArH), 7.12(bs, 3H, ArH and one ArOH), 7.28(s, 2 H, ArOH), 7.32(s, 1 H, ArOH). MS(FAB): 1175(MH+, 40%), Anal. calcd for C74H96O10S: C, 75.64; H, 8.18%. Found: C, 75.30; H, 8.24%.
REFERENCES
[1] Liu Y, Zhang H Y, You C C. Supramolecular Chemistry, Tianjin University Press, 2002.
[2] Ikeda A, Shinkai S. Chem. Rev., 1997, 97: 1713.
[3] Chen Y K, Chen Y Y. Chem. J. Chin. Univ., 1999, 10: 1570.
[4] Huang Z T, Zheng Y S. Chin. Chem. Lett., 1997, 8(8): 656.
[5] Li J S. Ph. D. Thesis, Wuhan University, Wuhan, 2001.
新型杯
[4]硫杂冠醚和杯[6]硫杂冠醚的直接合成杨发福 郭红玉 林建荣 (福建师范大学化学与材料学院, 福州 350007)
摘要 分别通过杯[4]芳烃和杯[6]芳烃与硫杂二甘醇双氯乙酸酯反应,用直接法合成了新型杯[4]硫杂冠醚3和首例杯[6]硫杂冠醚4, 产率中等。
关键词 杯芳冠醚, 硫杂, 合成