http://www.chemistrymag.org/cji/2005/077047ne.htm |
Jul. 2, 2005 Vol.7 No.7 P.47 Copyright |
A practical synthesis of dodecylmaltoside
Wang Yu, Fang
Zhijie*, Ye Lei ,Wang Yuanxing
(Chemical Engineering School, Nanjing
University of Science & Technology, Nanjing,210094)
Received Apr. 30, 2005; Supported by the
National Natural Science Foundation of China (No.29972020)
Abstract Hepta-O-acetyl-
a-D-dodecylmaltoside 2 had been achieved by Koenigs-Knorr reaction using hepta-O-acetyl-a-D-maltosyl bromide 1 which could be prepared by two-step method or by a novel one-pot process with maltose and lauryl alcohol as raw materials, then it was to give b-dodecylmaltoside 3 by deacetylation. The correlation experiments showed, the yield of compound 1 by one-pot process was 81% and enhanced 14% than by two-step process, it not only cut down to one step but also simplified the after-treatment, which will be convenient to be amplified in industry.Keywords Dodecylmaltoside, One-pot process, Koenigs-Knorr reaction
1. INTRODUCTION
b-Dodecylmaltoside as a
novel non-ionizing surface-active substances is more fitted to dissolve biomolecules than
higher alkylglucosides which are widely applied to medicaments and biological agents for
their bio-compatibility, high biodegradation and hypotoxicity etc, besides it improves
most effectively fold back effect not only for rerhodanase but also for dehydrogenase than
other surfactants[1-5]. The study on its pharmacological action has become hot
topic and its wide application has promising prospect.
A practical synthesis of dodecylmaltoside based on the route (Scheme 1)
with maltose as raw materials has been studied, which has two committed steps, one was for
synthesizing hepta-O-acetyl-
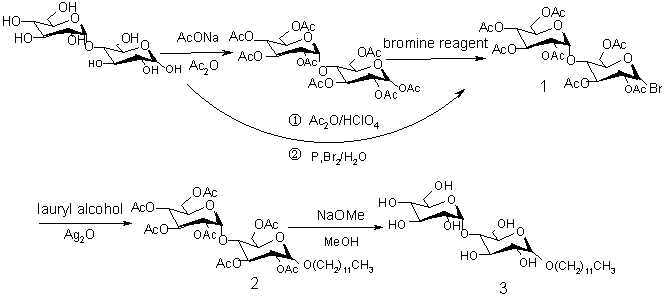
Scheme 1 The route of synthesizing b-dodecylmaltoside
2. EXPERIMENTAL
2.1 General Methods
Reactions were monitored by thin-layer chromatography (TLC)(silica gel HSGF254 glass
sheets).[
2.2 Preparation of hepta-O-acetyl-a-D-maltosyl bromide 1
Two-step method:
The synthesis of bromine reagent[6,7]: amorphous phosphorus (9g, 290.7mmol) was added slowly to 90ml glacial acetic acid, then it was stirred at 0ºC for 10 minutes. The solution was kept standing for 30min after 18ml bromine was dropped in, black precipitate was filtered to give yellow bromine reagent, which should be kept at 0ºC.
Sodium acetate (8.0g,96mmol) was added to 80ml acetic anhydride, the solution was boiled and stirred, dry maltose (10.0g, 56mmol) was added slowly and dissolved completely. The mixture was stirred at 100ºC and in a state of flux for 2h, then poured into 200ml ice-water, extracted with chloroform(50ml×3) to give the organic phase. It was respectively washed with water, NaHCO3 (100ml,5%) and water to give neutral solution, dried with anhydrous Na2SO4, filtered, and concentrated to give a yellow syrup, which was recrystallized from ethyl ether to yield 19.6g white crystal 1,2,3,6-tetra-O-acetyl-4-O-(2,3,4,6-tetra-O-acetylhexopyranosyl) hexopyranose (98.9%); mp:169ºC;
After 10ml bromine reagent was dropped slowly into 1,2,3,6-tetra-O-acetyl-4-O-(2,3,4,6-tetra-O-acetylhexopyranosyl)hexopyranose (3.5g,5.0mml), the solution was stirred for another 3h at 25ºC, then diluted with 50ml chloroform, washed respectively with ice-water (200ml×2) , NaHCO3 (100ml,5%) and 100ml water to give neutral solution, the flavescent solution was dried with anhydrous Na2SO4, filtered, and concentrated to give a yellow syrup, which was recrystallized from ethyl ether to yield 2.9g white power 1 (67%). mp:113ºC (lit.[9],mp:112-113ºC).
One-pot process:
0.2ml perchloric acid was dropped into 50ml acetic anhydride, the solution was stirred at 0ºC for 10 minutes and then heated to 30ºC. it was stirred for 2h at 38ºC after dry maltose (10.0g,28mmol) was added and dissolved completely, and cooled to 15ºC, amorphous phosphorus (3g, 96.9mmol) was added slowly, 6ml bromine was also dropped in batches, the solution was stirred for 3h and diluted with 50ml chloroform. It was filtered and washed with 100ml ice-water and 30ml chloroform respectively to give flavescent organic phase which would be extracted with saturated sodium hydrogen carbonate(200ml×8)to give neutral solution. The solution was dried with anhydrous magnesium sulfate, filtered and concentrated to a yellow syrup, which should be ground with ethyl ether to achieve 20.2g white powder 1(81%). mp:113ºC (lit.[9],mp:112-113ºC)。
2.3 Preparation of hepta-O-acetyl-a-D-dodecylmaltoside 2
A mixture of the lauryl alcohol(5.5902g,30mmol), hepta-O-acetyl-a-D-maltosyl bromide 1(2.8691g,4mmol), 40ml absolute ether and dry silver oxide (1.1587g,5mmol) in a tightly stoppered flask was shaken for one day, the ether solution was filtered through a thin layer of diatomaceous earth on a Buchner funnel, the ether evaporated and the excess alcohol removed by steam distillation upon cooling the residue in the flask, the crude heptaacetylmaltoside congealed and was filtered and crystallized from diethyl ether to give white power, column chromatography (1:1 petroleum ether-acetic ether) of the white powder gave 6.4g 2 (62%) ,mp:174ºC。
2.4 Preparation of b-dodecylmaltoside 3
To the solution of hepta-O-acetyl-b-D-dodecylmaltoside 2(3.0g,2mmol) in 30ml absolute methyl alcohol was added 0.1N solution of sodium methylate in 2ml methyl alcohol. The solution was boiled for two hours under a reflux condenser protected by a calcium chloride tube. The mixture was concentrated with the reduced pressure distillation to give a yellow syrup. The syrupy residue was taken up in diethyl ether and was filtered to give white power 1.7g 3(83%).mp:240ºC;NMR(DMSO+TMS): d 8.48(s,1H, H-1);d 4.99(s,2H,H-5/-5');d 4.13(d,2H,H-4/-4');d 3.73(d, 2H, H-2/-2');d 3.58(s,2H,H-3/-3');d 3.06-3.44(m,12H,OH-2,-3, -6/-2', -3',-4', -6',H-6/-1', -1');d 1.06-2.50 (m,22H,-CH2);d 0.86(m,3H,-CH3). ESI-MS(m/z)(M=510) : 474.9(M-2OH) , 492.9(M-OH),508.9(M-H),509.9(M),532.9(M+Na),534.0(M+Na+H),573.9(M+K+Na).
3. RESULTS AND DISCUSSION
The developed novel one-pot process to achieve acetylated maltosyl bromide 1 as
donor of disaccharide groups was more convenient and efficient than the two-steps method
based on the route generally used for acetylating monosaccharide [9-13], the
purity and yield was improved.
The glucosidic bond was formed effectively, which was based on the
Koenigs-Knorr reaction widely applied to monosaccharide[14-16].With silver
oxide as catalyst, the reaction had twelvefold time, it was two hours for monosaccharide
having lower steric hindrance and higher activity [15,16], the products frequently came down as a milky emulsion
which crystallized on cooling in an ice-bath, the yield varied from 45 to 62% of the
calculated amount.
The deacetylation of hepta-O-acetyl-a-D-dodecylmaltoside 2 was also based on the widely
used method for acetylalkylglucoside whose reaction time is one-half hour
[11,12].It is incomplete, diethyl ether can
dissolve fully unreacted material to give straight product, the extraction is simpler and
more effective than the latter's whose product was crystallized from water or ethyl
acetate, the extract yield was 100%.
More and more alkylpolyglucosides(APG)with glucose as raw material have been extensively studied and
widely used, while there is little study on high purity alkyldisaccharosides with
disaccharide as raw material. Along with our research achievements of dodecylmaltoside, we
will further study the property and function of it to do truly make extensive use in
biomedicine fields, such as a medicine absorption enhancer or biosolvent for
biomacromolecule et al[17,18].
REFERENCES
[1] Joanne L M, Reithmeier R A F, Wang Daneng. Journal of Structural Biology, 2002,
137 (3): 322-332.
[2] Ke T L, Cagle G, Schlech B et al. Journal of Ocular Pharmacology and Therapeutics,
2001, 17 (6): 555-563.
[3] Ferrer M, Comelles F, Plou F J,Cruces M A et al. J. Am. Chem. Soc. 2002, 18 (3):
667-673.
[4] Fan B,RosenB P. Journal of Biological
Chemistry,2002, 277 (49): 46987-46992.
[5] Scheffel F, Fleischer R, Schneider E. Biochimica et Biophysica Acta, 2004, 1656 (1):
57-65.
[6] Zhu Qianjin. NanJing University of Science and Technology Master Dissertation, 2004,
30-31.
[7] Sun J L, Mi S, Wang W T. Chem Bull, 2000, (3): 45.
[8] Zhao Chengfang. The Synthesis and Application of Alkylpolyglycoside Derivate. The
Master Degree Dissertation of Nanjing University of Science & Technology, 2001, 29.
[9] Haynes L J, Newth F H. In Advances in Carbohydrate Chemistry, 1955, 10: 207-210.
[10] Wolfrom,M L, Thompson A. In Methods in Carbohydrate Chemistry, 1963, 2: 211-216.
[11] Redemann C E, Niemann,C. Org Synth.1942, 22 (1): 314-319.
[12] Kartha K P R, Jennings H J. J Carbohydr Chem.1990, 15: 955-959.
[13] Randazzo G, Capasso R, Rosaria M et al. Carbohydrate Research. 1980, 85: 298-301.
[14] Li Yuwen, Li Yingxia, Zhang Wei. Chinese Journal of Organic Chemistry(You Ji Hua
Xue).2004, 24 (4): 438-439.
[15] Noller C R, Rockwell W C. J Am Chem Soc.1938, 60: 2076-2077.
[16] Lian Zhaobin, Huan Yan, Cao Linghua. Chinese Journal of Applied Chemistry (Ying Yong
Hua Xue), 2003, 20 (3): 287-288.
[17] Ke T L, Cagle G, Schlech B et al. Journal of Ocular Pharmacology and Therapeutics,
2001,17 (6):.555-563.
[18] Leopold L L, Iban U B, Christopher G T et al. J Am Chem Soc, 2004, 126 (44):
14362-14363.
十二烷基麦芽糖糖苷的实用性合成
王煜,方志杰*,叶磊,王远兴
(南京理工大学化工学院,南京,210094)
摘要 以麦芽糖和月桂醇为原料, 通过两步法或一锅法合成七-O-乙酰基-a-D-溴代麦芽糖1,后者经Koenigs-Knorr反应得七-O-乙酰基-a-D-麦芽糖糖苷2,再脱乙酰基得b-十二烷基麦芽糖糖苷3。对比实验表明:一锅法制取活性中间体1的得率达81%,较两步法提高了14%,不但减少了一步反应,而且简化了后处理操作,便于工业放大。
关键词 十二烷基麦芽糖糖苷,一锅法,Koenigs-Knorr 反应