http://www.chemistrymag.org/cji/2006/081004pc.htm |
Jan. 10, 2006 Vol.8 No.1 P.4 Copyright |
Chen Shaoyuan1,2, Yang Jun1,2*, Tang Yu1,2,Zhang Yuanming1,2
(1 Department of Chemistry, Jinan University; 2 Institute of
Nanochemistry, Jinan University, 510632, China)
Abstract Anatase TiO2 nanoparticles were prepared by direct decomposition of Ti(SO4)2. The effect of calcination temperature on the crystalline phase, physical and chemical properties of the resulting particles were also investigated. The results showed that the calcination temperature had dominant influence on crystalline phase, specific surface area and photocatalytic performance of the TiO2 particles. At a temperature below 600
ºC, the TiO2 crystalline phase in high order could not be obtained and anatase phase appeared at 600ºC, in which TiOSO4 was still the major composition. When the temperature reached 700ºC, the sample completely transformed to anatase phase with a crystalline size of 18 nm, As the temperature further increased to 800ºC and 900ºC, the specific surface area of the catalyst decreased sharply. However, only the anatase phase was present. This strongly suggested that the presence of SO42- restrained the transformation of anatase into rutile and strengthened the ability against sintering. The catalyst calcined at 700ºC showed the optimal catalytic performance and the highest specific surface area. The percentage of methyl orange decoloration at 90 min reached 99.9 %.Keywords titania; photocatalysis; titanium sulfate; decomposition Ti(SO4)2热分解法制备锐钛矿型纳米TiO2及光催化研究
陈绍源
1,2,杨骏1,2*,唐渝1,2,张渊明1,2(1 暨南大学化学系;2 暨南大学纳米化学研究所,广州,510632)
2005年12月8日收稿;国家自然科学基金863计划资助项目(2001AA523010);广东省自然科学基金团队项目(05200555).
摘要 采用
Ti(SO4)2直接加热分解制备了锐钛矿型纳米TiO2,结果表明:焙烧温度对样品的晶相、比表面和光催化性能有决定性的影响。600 ℃以下温度焙烧不能获得规整的TiO2晶相,700 ℃时完全转变为锐钛矿相TiO2微晶,晶粒大小约18 nm;继续升高温度,催化剂的比表面因晶粒变大而急剧减小,但由于硫酸根离子(SO42-)的抑制作用,900 ℃时,样品仍然保持单一锐钛矿相。700 ℃焙烧的样品具有最大的比表面和最高的光催化活性,光催化降解甲基橙溶液90 min,降解率为99.9 %。关键词 二氧化钛;光催化;硫酸钛;热分解法
1 引言
自从1972年Fujishima和Honda发表了关于氧化钛电极上光分解水的论文以来,TiO2作为一种光催化剂越来越受人们的广泛重视[1]。纳米二氧化钛是一种新型的无机功能材料,它具有许多独特的性质,如化学活性高、可见光透过性好、吸收紫外光性能强,具有超亲水性等[2-3],可用于制造电介质材料[4]、光催化薄膜[5-6]、化学传感器等[7]。TiO2作为光催化剂,与其它催化剂相比,TiO2具有来源丰富、光照后不发生光腐蚀、耐酸碱性好、化学性质稳定和无毒等优点,因而是当前最具应用潜力的一种光催化剂。由于TiO2受光激发后产生的光生电子和空穴具有高的电势电位,有很强的氧化性和还原性,作为光催化剂几乎能将水体和空气中的绝大多数有机污染物最终降解为H2O、CO2和无害盐类。TiO2主要有两种晶型:锐钛矿型和金红石型。两种晶型都是由相互联接的TiO6八面体构成。两者差别在于八面体的畸变程度和八面体间相互联接的方式不同。金红石型八面体不规则,微显斜方晶畸变,对O2的吸附能力较差,比表面积较小,光生电子和空穴容易复合,催化活性受到一定影响;而锐钛矿型八面体呈明显的斜方晶畸变,其对称性较前者低,光催化活性较高。在一定温度处理下,锐钛晶型TiO2可以转化为金红石型,导致TiO2光催化活性的降低。
制备纳米TiO2的方法很多,有物理法和化学法[2, 8-10]。物理法常用的有构筑法(如气相冷凝)和粉碎法(如高能球磨法)等。化学法主要有沉淀法、水解法、喷雾热解法、溶胶-凝胶法、氧化还原法和水热法等。这些方法中多采用钛醇盐、钛酸酯和TiCl4等为原料,涉及硫酸钛的研究还很少,对于硫酸钛为原料的制备法的深入研究尤其缺乏。最近,有研究者[11-12]采用硫酸作用下硫酸钛溶液直接强迫水解制得了纳米尺度的TiO2,此催化剂具有高的苯酚降解光催化活性。但是,在他们的方法中需对沉淀进行多次过滤洗涤,由于TiO2的颗粒很细,这不仅耗时且降低了TiO2的收率。
成本低,操作简易地制备高催化性能的纳米二氧化钛材料是现阶段人们努力的方向。本文选择Ti(SO4)2作原料,采用热分解法制备了锐钛矿型纳米TiO2粉体。该法极其简便易行,TiO2的收率也高,且样品经900 ℃高温焙烧后仍保持单一的锐钛矿晶相。
2 实验部分
2.1 实验试剂
实验所用的试剂为:硫酸钛(CP上海润捷化学试剂有限公司),甲基橙(AR湖南金涛化学试剂研究所)
2.2 实验仪器
GGZ-125W型高压汞灯(上海亚明灯泡厂125 W),可见分光光度计(Spectrumlab 22pc, Lengquang Tech公司),TDL-5型离心机(上海安亭科学仪器厂),DL-60A型超声波清洗器 (上海之信仪器有限公司),82-4-1J高温箱形电炉(上海博讯实业有限公司)。
2.3 TiO2的制备
称取一定量的Ti(SO4)2,在玛瑙研钵中研细到小于100目后,置于80-90 ℃的电热鼓风干燥箱内干燥2 h,最后放入马弗炉中,以5 ℃/min的升温速度分别升至在不同温度下焙烧2 h即可获得纳米TiO2样品。
2.4 TiO2的表征
X-射线衍射分析(XRD):在布莱格科技(北京)有限公司MSAL XD-2型X射线衍射仪上测定样品的物相,辐射源为Cu Ka,l= 0.15418 nm,衍射角2q为5-80°,工作电压40 kV,电流20 mA,扫描速度为8°/min,步宽为0.02°。
热重TG分析:在上海精密科学仪器有限公司ZRY-2P 型综合热分析仪上进行,空气氛下以10 ℃/min升温速率升至1000 ℃。
比表面积测定:低温N2吸脱附由Tristar 3000物理吸附仪(美国Micrometrics公司)测定,样品于120℃烘干后置于干燥器中冷却至室温,然后移至样品管,在温度200 ℃,压力10-2torr的条件下,处理样品4 h,液氮冷却至-196 ℃,进行低温N2吸脱附实验,数据由计算机自动采集。由Brunauer Emmett Teller(BET)方程计算样品的比表面积。
2.5 光催化活性测试
取配好的0.020 g/L甲基橙水溶液100 ml置于250 ml烧杯中,往其中加入0.050 g制得的催化剂样品,超声使其均匀分散后,转入配有125 W汞灯(上海亚明灯泡厂GGZ-125 W型,lmax=365 nm)的自制可控温光反应器中进行光降解性能测试。反应装置要求光源与液面垂直距离约12 cm,同时鼓入空气以提供反应所需氧气,空气由微型空气泵供给(90mL/min),反应时不断搅拌,反应温度控制在28 ℃左右。每隔一段时间(30 min)抽取一定量的溶液,经离心分离后取上层清液用Spectrumlab22pc分光光度计测量甲基橙溶液在最大吸收峰波长(约464 nm)处的吸光度A。
3 实验结果与讨论
3.1 TiO2的表征
3.1.1 XRD和TG分析
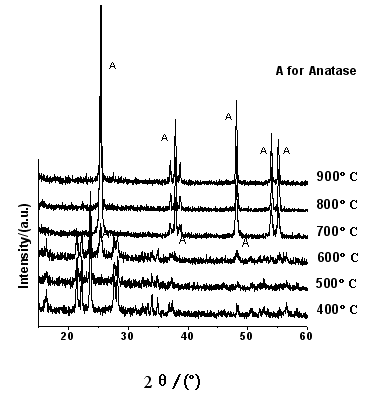
图1 不同热分解温度的TiO2的XRD图
Fig.1 XRD patterns for TiO2 particles calcined at different temperatures
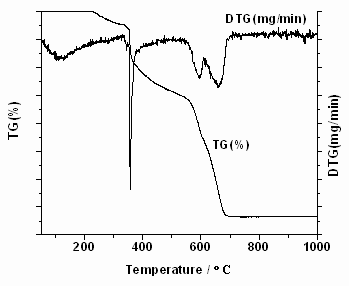
图2 Ti(SO4)2的TG-DTG曲线图
Fig 2 TG and DTG patterns of Ti(SO4)2
图1和图2分别是Ti(SO4)2经不同热分解温度处理后得到的TiO2粉体的XRD图和Ti(SO4)2的热重图。从图1可见,焙烧温度为400和500 ℃时,未出现TiO2的特征衍射峰,谱图上主要是TiOSO4的特征峰(2
q为21.3,23.5和28.0°);当焙烧温度达到600 ℃时开始出现锐钛矿的特征峰,但此时依然伴随着TiOSO4峰,从峰强度来看,TiOSO4含量仍占相当的比重。另外,从外观上看,400-600 ℃焙烧得到样品呈微黄色,有文献报道这是表面残留了SO3所致[13]。而从图2中DTG曲线上可以看到,200 ℃之前出现了一个峰,为脱表面吸附水峰;在300-400 ℃和600-700 ℃又出现两个峰,在其TG曲线上皆有对应的失重;在温度低于350 ℃,样品的失重率约为20 %;根据文献报道,如按Ti(SO4)2分子中含四分子结晶水算,所占比例为23 %[13],由此可推断350 ℃前的失重归因于样品的结晶水脱出;400-500 ℃区间Ti(SO4)2开始分解为TiOSO4,XRD图中出现对应的TiOSO4特征峰。图1中,当焙烧温度达到700 ℃时,硫酸氧钛的衍射峰消失,只有锐钛矿相TiO2特征峰存在,晶粒大小约18 nm。继续升温到 800 ℃和900 ℃时,TiO2仍保持单一的锐钛矿相,未见金红石相的特征峰出现,但此时衍射峰明显增强,表明样品的晶化程度增加,晶粒变大为约26 nm和40 nm。由TG可见,700 ℃时样品的失重比例约为74 %,已接近于含四分子结晶水的Ti(SO4)2的最大失重量,表明Ti(SO4)2已几乎完全分解转变为TiO2晶相,对应于样品XRD图中TiOSO4特征峰的消失。大量文献结果表明[2, 8, 14],温度是促进TiO2晶相转变的敏感参数,一般从无定形TiO2到锐钛矿的变化主要发生在400-500 ℃;550-700 ℃是锐钛矿相转变成金红石相的主要区间;当煅烧温度为750 ℃时,样品一般可全部转化为金红石相。武瑞涛等[11]采用Ti(SO4)2水解法制备TiO2,当焙烧温度为550 ℃时,已有部分锐钛矿相转变为金红石相;焙烧温度800 ℃时,TiO2已完全转化成金红石型相。而彭峰等[12]的研究中,焙烧温度600 ℃时,样品未出现金红石相,800 ℃焙烧样品才开始出现金红石相结构,而此时锐钛矿相仍是主要的组成(92.5 %)。与他们不同,本文制备的样品在900 ℃焙烧都未出现金红石型晶相。产生上述差异的原因可能是硫酸根离子(SO42-)存在所致。有文献报道[15]硫酸根离子SO42-对TiO2由锐钛矿型向金红石型的晶型转变起一定的抑制作用。Carp等[2]也报道对SO42-/TiO2固体超强酸,SO42-的加入提高了锐钛矿相的稳定性以及催化剂的抗烧结性能;与非硫酸化的TiO2相比,加SO42-的TiO2在700 ℃焙烧时仍保持锐钛矿相,且具有相对大的比表面。在武瑞涛[11]和彭峰[12]等的工作中,对沉淀进行了多次抽滤和洗涤,残留SO42-离子的数量不同,可能是导致他们所制样品金红石相出现温度不同的原因。而在笔者的实验过程中并没有对样品进行洗涤等步骤,因而在焙烧过程中存在大量的硫酸根离子SO42-,因而抑制了锐钛矿向金红石的转变,故900 ℃焙烧都未出现金红石型晶相。
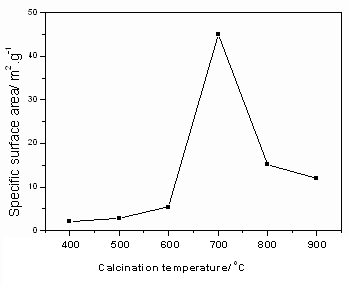
图3 焙烧温度对催化剂比表面的影响
Fig. 3 Effect of calcination temperature on specific surface area of catalysts
3.1.2 BET比表面和TEM
不同焙烧温度处理的TiO2样品的比表面示于图3。由图可见,焙烧温度对样品的比表面的影响显著,在低于600 ℃时,样品的比表面随温度的升高而缓慢地由400 ℃时的2.2升至600 ℃时的5.4 m2/g, 700 ℃时样品的比表面急剧升至45.0 m2/g,继续升高温度,样品比表面又开始急剧的减小,800 ℃焙烧样品比表面15.2 m2/g。由前面的XRD和TG结果可知,400-500 ℃的低比表面是因为TiOSO4未分解;600 ℃TiOSO4开始分解,样品的比表面明显由500 ℃时的2.8升至600 ℃时的5.4 m2/g,温度升到700 ℃时,TiOSO4已完全分解,对应的样品具有最大的比表面45.0 m2/g;继续升高温度,使得TiO2晶粒增大,致使其比表面明显降低。
700 ℃焙烧样品的TEM图示于图4。由图可见,样品呈球形颗粒,且粒径分布均匀,约为25-30 nm,大于由XRD图谱经谢乐(Scherrer)公式计算的晶粒大小,两者不一致的原因是TEM显示的是TiO2颗粒大小,而颗粒往往是由更小的晶粒构成的。
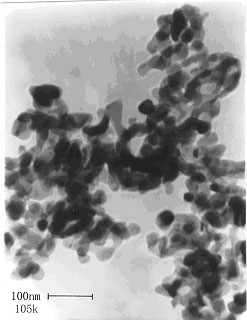
图4 700
Fig. 4 TEM of catalyst calcinated at 700 ℃
3.2 TiO2催化剂的光催化测试分析
图5是0.020 g/L甲基橙水溶液在高压汞灯照射下,甲基橙的剩余百分率与TiO2焙烧温度之间的关系;此外,比表面积最大的700 ℃焙烧样品在无光照情况下对甲基橙的吸附曲线也列于图5中。由图可见,焙烧温度对催化剂光催化性能有很大的影响,未加催化剂的情况下,甲基橙的降解程度不明显。低于700 ℃时,催化剂的活性随焙烧温度的升高而升高,400和500 ℃焙烧的催化剂主要是TiOSO4,对应低的光催化活性,降解率分别28.4和37.5 %;600 ℃开始出现锐钛矿相的TiO2,其降解率升高到45.6 %;700 ℃焙烧时,样品已完全转变为锐钛矿相的TiO2,且具有最大的比表面45.0 m2/g,对应最优的活性,降解率为99.9
%,但在无光照下该样品对甲基橙的吸附量极小;继续升高温度,虽然样品仍然保持锐钛矿相,但比表面急剧降低,因而光催化活性也分别降低到800和900 ℃的87.5和85.1 %。虽然800和900 ℃焙烧的催化剂比表面急剧由700 ℃时的45.0 m2/g降至15.2和11.9 m2/g,但活性降低的程度远低于催化剂比表面降低的程度,这表明比表面并非决定催化剂活性的唯一重要因素,催化剂的晶相和形貌等也有很大的影响。本文中的催化剂由于采用Ti(SO4)2直接热分解制备,焙烧过程中大量存在的硫酸根离子(SO42-)存在抑制了锐钛矿向金红石的转变,使得样品经900 ℃焙烧仍保持单一的锐钛矿相,由于锐钛矿相具有高的光催化活性,因而催化剂经高温焙烧后仍有较好的光催化活性。
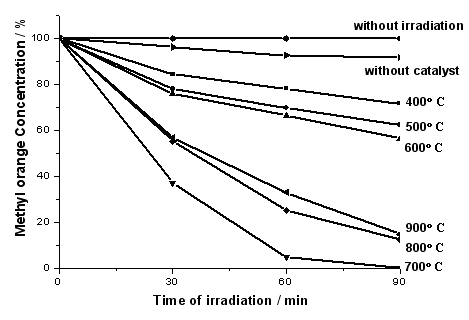
图5 不同温度焙烧的TiO2的甲基橙光催化降解性能
Fig. 5 The
photocatalytic performance of catalysts calcinated at different temperatures for
degradation of methyl orange
4 结语
以Ti(SO4)2作原料直接热分解法可方便、快速的制备锐钛矿型纳米TiO2粉体。焙烧温度对样品的晶相、比表面和光催化性能有决定性的影响。600
℃以下温度焙烧不能获得规整的TiO2晶相,400-500 ℃为硫酸氧钛,600
℃开始出现锐钛矿相TiO2,但此时催化剂仍以硫酸氧钛为主;700 ℃焙烧时样品完全转变为锐钛矿相TiO2微晶,晶粒大小约18 nm;继续升高温度,催化剂的比表面因晶粒变大而急剧减小,但到900
℃时,样品仍然保持单一锐钛矿相。由此可推断,硫酸根离子(SO42-)存在抑制了锐钛矿向金红石的转变,增加催化剂的抗烧结性能,因而,样品经700
℃焙烧后仍为锐钛矿相且具有相对高的比表面,同时其光催化活性最高,光催化降解甲基橙溶液90
min,降解率为99.9 %。
REFERENCES
[1] Fujishima A, Honda A. Nature, 1972, 238, 37.
[2] Carp O, Huisman C L, Reller A. Progress in Solid State Chemistry, 2004, 32(1-2):
33-177.
[3] Calza P, Medana C, Pazzi M et al. Applied Catalysis B: Environmental, 2004, 53(1):
63-69.
[4] Wen X M, Xie S W, Lin L Z. Acta Physica Sinica (Wuli Xuebao), 1997, 46(8): 1652-1657.
[5] Ostomel T A, Stucky G D. Chemical Communications, 2004, 1016-1017.
[6] Zhao L L, Yu Y, Song L X et al. Applied Catalysis A-General, 2004, 263(2): 171-177.
[7] Ruiz A M, Sakai G; Cornet A et al. Sensors and Actuators B, 2004, 103(1-2): 312-317.
[8] Gao L, Zheng S, Zhang Q H. Photocatalytic Materials of Nano-titanium Oxide and their
Applications (Na'mi Yanghuatai Guangcuihua Cailiao Ji Yingyong). Beijing: Chem Ind Press,
2003.
[9] Tanaka Y, Suganuma M. Journal of Sol-Gel Science and Technology, 2001, 22(1/2): 83-89.
[10] Zhang W, Xie H Y. Chemical Equipment Technology (Huagong Zhuangbei Jishu), 2004,
25(4): 43-46.
[11] Wu R T, Wei Y. Journal of Inorganic Materials (Wuji Cailiao Xuebao), 1996,14(3):
461-464.
[12] Peng F, Ren R Q, Lei J G. Modern Chemical Industry (Xiandai Huagong), 2002, 22(S1):
108-110.
[13] Liao D Z, He J Y, Zhong M. Chemistry (Huaxue Tongbao), 2003(5): 346-350.
[14] Liu H Z, Chen H Y, Zhang H et al. Journal of Shanghai Jiaotong University (Shanghai
Jiaotongdaxue Xuebao), 2001, 35(5): 680-683.
[15] Li Y Z, Lee N H , Hwang D S et al. Langmuir, 2004, 20(25): 10838-10844.