http://www.chemistrymag.org/cji/2006/081007pc.htm |
Jan. 10, 2006 Vol.8 No.1 P.7 Copyright |
(Department of Chemistry and Chemical Engineering, Weinan Teacher's College,Weinan 714000,China) Abstract A new type of polarographic catalytic wave of organic compound, termed as association / parallel catalytic hydrogen wave, has been reported. The Association / parallel catalytic hydrogen wave with such characters as the negative shift of the peak potential and the great increase of the peak current in famotidine - persulfate system was studied. Here persulfate as ligand formed an association complex with protonated famotidine, which caused the peak potential of catalytic hydrogen wave of famoditne to shift negatively. Additionally, the great increase of the peak current was due to the further catalysis of the catalytic hydrogen wave by sulfate radical produced in the reduction of persulfate at electrode. In 0.1mol/L KH2PO4-Na2HPO4 (pH 6.2) -2.0��10-5mol/L K2S2O8 supporting electrolyte , famotidine yielded a association/parallel catalytic hydrogen wave with a peak potential of Ep-1.42V��vs.SCE��. The 2nd-order derivative peak currents (ip") of the wave varied linearly with the famotidine concentration over the range 4.0��10-7 to��6.0��10-5 mol/L (r = 0.9991, n = 9) and 5.0��10-8 to 4.0��10-7mol/ L (r = 0.9989,n = 8) with detection limit of 1.5��10-8 mol/L (3s). RSD (n = 13) was 1.32% at the concentration level of 8.0��10-7 mol/L the analytical sensitivity of the association / parallel catalytic wave was 10 times higher than that of the corresponding catalytic wave. The proposed method has been applied to the determination of famotidine in tablets without previous treatment.
Keywords Famotidine, potassium persulfate , catalytic hydrogen wave, association/parallel catalytic hydrogen wave
��
��Ī�涡��K2S2O8 ��/ƽ�д��Ⲩ���ⶨ�о� ������ ��������μ��ʦ��ѧԺ��ѧ����ϵ������ μ�� 714000�� ����ʡ������ר����л��� (03JK216) ������Ŀ ժҪ���������л������K�״�����һ��������----��/ƽ�д��Ⲩ���о���Ī�涡��FMT����K2S2O8�ĵ�/ƽ�д��Ⲩ�Ľ�����������ӻ�FMT��S2O82-�γɵ��������λ���ƣ�FMT���Ⲩ��S2O82-��ԭ�м����SO4-·��һ������ʹ������������ӡ���0.1mol/L KH2PO4-Na2HPO4��pH 6.2��-2.0��10-5mol/L K2S2O8��֧�ֵ�����У�FMT��EP = -1.42V (vs.SCE)�������ĵ�/ƽ�д��Ⲩ����������������Ũ����4.0��10-7 ~ 6.0��10-5 mol/L��r = 0.999 1, n = 9����5.0��10-8 ~ 4.0��10-7 mol/L(r = 0.998 9,n = 8����Χ�ڳ����õ����ι�ϵ�������Ϊ1.5��10-8 mol/L(3s)��13��ƽ�вⶨ2.0 ��10-7 mol/L��FMT����Ա�ƫ��Ϊ1.32%���õ�/ƽ�д��Ⲩ����Ӧ�Ĵ��Ⲩ��߷���������10�����÷�������Ҫ��ƷԤ��������ֱ�ӲⶨƬ����FMT�ĺ�����
�ؼ��ʡ���Ī�涡��K2S2O8�����Ⲩ����/ƽ�д��Ⲩ ��Ī�涡(Famotidine��FMT��Ϊһ����ĸ�˲����Ͼ��ж�������ļ��Ի���������Խ��������ܽ⣬��ˮ���״�����ˮ�Ҵ��Ƚ����м��ܡ���ѧ��Ϊ3-[[[2-[���������Ǽ�������]-4-�����]��]���]-N-�����������ߡ�FMT�Ǽ������涡�������涡֮���Ƴ��ĵ���������H2 ���������ͼ���ͨ������θ��ϸ����H2���壬����θ����ڶ���Ч����θ��ʮ��ָ������[1]������ҩ��1995�����ص��ʿط����ǣ�ԭ��ҩ���ø�����ζ���Ƭ�����ø�ЧҺ��ɫ����귨��������Ϊ�״�-ˮ-�����λ���Һ(6:31:3)���ڹ轺���Ͻ���[2]���ҹ��������÷�ˮ�ζ����ⶨԭ�ϣ�����ֹ��ȷ��ⶨƬ��[3]���й�FMT�ⶨ�����������ױ�������Ҫ��HPLC��[4-6]��RH-HPLC[7-8]������ֹ��ȷ�[9-10]���绯ѧ��[11,12]��Squella��[11,12] ���ڴ��Ⲩ���ཻ�������ⶨ��FMT�ĺ���������FMT�ڲ�̼�缫�������ò�����弫���ⶨ��FMT�ĺ����������ʴﵽ99.1%��
�л�������ļ��״����ɷ�Ϊ�л�������ɻ�ԭ���ŵ�ƽ�д���[13-15]�����ƽ�д��������ƽ�д�������������������ԭ���Ⲩ����һ�����������ģ���ʵ�����л���������Я�������ӻ�ԭ�м���ԭ��̬�ⱻ�������������������ӣ��������Ƿ�������ӣ����λ����[16,17]���ο����[18]������H2O2�Ĵ����£���������������ø�ش��Ⲩ�Ĵ����·�������ӣ����λ���ƣ���֮Ϊ����/ƽ�д��Ⲩ�������߷�����KH2PO4-Na2HPO4��pH 6.2������Һ�У�FMT�ڷ��λ-1.38V�������Ĵ��Ⲩ����K2S2O8�����£���������ӣ����λ������-1.42V����������FMT�ĵ�/ƽ�д��Ⲩ���������������ip"ֵ��4.0��10-7 ~ 6.0��10-5 mol/L��5.0��10-8 ~ 4.0��10-7 mol/L��Χ�ڳ����õ����Թ�ϵ�����ô˷������ԲⶨFMT�ĺ�������ҩ�����кܺõ�ʹ�ü�ֵ�� 1 ʵ�鲿��
1.1�������Լ�
JP-303��ʾ�������ǣ��Ĵ��ɶ��������������缫ϵͳ���ι��缫Ϊ�����缫������ɨ�裨303A�����缫Ϊ�����缫��ѭ�������о��������ʹ��缫Ϊ�αȵ缫�����缫Ϊ�����缫��
1.0��10-3mol/L FMT������Һ�����ƣ�ȷ��ȡ0.168 5g FMT�����ݱ���ҩҵ����˾��������99.0��������ϡ�������ܽ⡢���ˡ���ˮ������500mL����ƿ�С�������Һ��ˮϡ�͵�����Ũ�ȡ�FMT��Ʒ�IJⶨҺ����Һ�����Ʒ����������Լ���Ϊ������,ʵ����ˮ��Ϊ��������ˮ��
1.2 ʵ�鷽��
ȷ��ȡһϵ��FMT��������Һ1.0mL��25mL������ƿ�У�����2.0��10-4mol/L K2S2O8��Һ 2.5mL��0.2mol/L KH2PO4��Һ 10.0mL, 0.2mol/L Na2HPO4��Һ 2.5mL (pH 6.2������Һ��Ũ��Ϊ0.1mol/L)����ˮϡ�����̶ȣ�ҡ�ȣ�ת������������Һ��10mL���׳��У���JP-303�ͼ�����������������ɨ�裬��ʼ��λΪ-0.90 V��ɨ������Ϊ0.25V·s-1������FMT��-1.42V (vs. SCE) �����״����Ķ�����������ip�� ֵ����FMT��Ũ�ȺͶ�����������ip�� ֵ�������Իع鷽���� 2 ���������
2.1��ʵ���������Ż�
2.1.1֧�ֵ��������������ѡ��
ʹ��JP-303��ʾ����������������ɨ�裬 ��-0.90 ~ -1.90V��λ��Χ�ڡ�������FMT��HAc-NaAc��KH2PO4-Na2HPO4��NH4Cl-NH3·H2O��KH2PO4- Na2B4O7��KH2PO4-NaOH��Britton-Robinson��KCl��NaClO4��NaOH��HCl��H2SO4�ȶ���֧�ֵ���ʵļ�����Ϊ�������������HAc-NaAC��Na2HPO4-KH2PO4��NH4Cl-NH3·H2O����Һ�У�FMT���������Ⲩ���ֱ������ͬŨ�ȵ�K2S2O8��H2O2��KIO3�������ô��Ⲩ����һ������������������λ���ơ���K2S2O8��FMT�Ĵ��Ⲩ�Ĵ����������ԣ������ĵ�/ƽ�д��Ⲩ�ķ������������ζԳ����ȶ�(ͼ1,d��)����ˣ�����ѡȡKH2PO4-Na2HPO4����Һ��Ϊ֧�ֵ���ʣ� K2S2O8��Ϊ��������
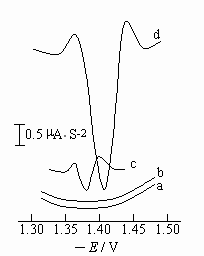
ͼ1 FMT�Ķ���������ͼ
a 0.10mol/L NaH2PO4-K2HPO4 (pH6.2) b a + 2.0��10-5mol/L K2S2O8
c a + 2.0��10-7mol/L FMT d b + 2.0��10-7mol/L FMT
2.1.2������ҺpHֵ��Ũ�ȵ�Ӱ��
������KH2PO4-Na2HPO4����ҺpHֵ�仯��FMT��/ƽ�д��Ⲩ�����ip"ֵ�����λEp��Ӱ�졣�����������pH��4.5 ~ 6.2��Χ�ڣ� ip"ֵ�滺��ҺpHֵ���߶�������Ep���ƣ�pHֵ��6.2-8.0��Χ�ڱ仯ʱ, ip"ֵ��С������ѡȡpHֵΪ6.2��KH2PO4-Na2HPO4��Ϊ����Һ��
ά��KH2PO4-Na2HPO4����ҺpH 6.2�㶨��������Һ��Ũ����0.5��10-3 ~ 0.15mol/L��Χ�ڱ仯ʱ��Ep���ƣ�����Ũ�ȴ�0.3��10-3 mol/L������1. 0��10-2mol/Lʱ�� ip"ֵͻԾ������Ũ����1.0��10-2 ~ 0.10mol/L��Χ�ڣ� ip"ֵ�������ӽ�һƽ̨������Ũ����0.10 ~ 0.15mol/L��Χ�ڣ� ip"ֵ������С����Ũ��Ϊ0.10mol/L����ʱ�����λEp -1.42V��vs.SCE����ip"ֵ����ζԳ����ȶ����ʱ���ѡȡKH2PO4-Na2HPO4����Һ��pH 6.2������Ũ��Ϊ0.10mol/L��
2.1.3 ��K2S2O8Ũ�ȵ�Ӱ��
��pH6.2��KH2PO4-Na2HPO4Ϊ����Һ�У�����K2S2O8Ũ�ȴ�2.0��10-7mol/L���ӵ�3.0��10-5mol/L��FMTƽ�д��Ⲩ�����ip"ֵͻԾ������K2S2O8Ũ�ȴ�3.0��10-5mol/L���ӵ�2.0��10-4mol/L��ip"ֵ������С��ͼ3������ˣ�����ѡȡ������K2S2O8��Ũ��Ϊ2.0��10-5mol/L��
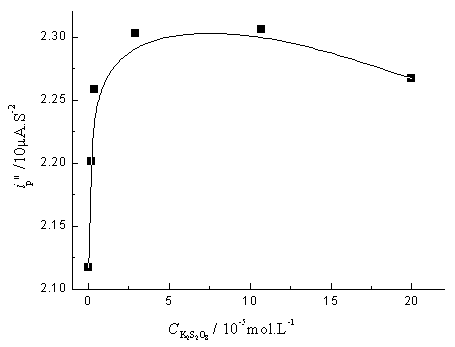
Fig.2 Effect of K2S2O8 concentration on peak current of 2.0��10-7mol/L FMT in 0.10mol/L KH2PO4-Na2HPO4 (pH6.2) buffer ��������������ѡ��0.10mol/L KH2PO4-Na2HPO4����Һ��pH 6.2������Ϊ���֧�ֵ���ʣ�2.0��10-5mol/L K2S2O8��Һ��Ϊ�����������ö���������������ʼ��λΪ-0.90V����������ɨ�裬�ⶨFMTƽ�д��Ⲩ�Ķ�����������ip��ֵ��
2.2 �ȶ��Լ�������
ʹ��JP-303��ʾ����������������ɨ�裬ʵ����������2.0 ��10-7mol/L��Һ���ƺ�12h�ڣ�FTMƽ�д��Ⲩ�Ķ�����������ip"�ͷ��λEp�������䣬�ȶ������á�13��ƽ�в���2.0��10-7mol/L FTMƽip��ֵ����Ա�ƫ��RSDΪ1.32%��
2.3 ���������
�ڱ�ʵ����ѡ������ѵ�Һ�У�FTMƽ�д��Ⲩ������������ip����nA·S-2��ֵ����Ũ����6.0��10-7 ~ 6.0��10-5mol/L��5.0��10-8 ~ 4.0��10-7mol/L��Χ�ڳ����õ����Թ�ϵ�������Իع鷽�̷ֱ�Ϊip" = -11.04 + 1.34 ��107CFTM��r = 0.999 1, n = 9���� ip" = 9.99 + 1.68 ��105CFTM (r = 0.998 9, n = 8���������Ϊ1.5��10-8mol/L (3s)��
2.4 �������ʵ�Ӱ��
������һЩ���ܲ�������FTM�ĸ��ͼ���Ƭ���е����϶�2.0��10-7 mol /L FMTƽ�д��Ⲩ��Ӱ�졣����������������������5%��Χ�ڡ����ۣ����������ǣ��ȼ������ƣ��DZ�����ά�أ���ʯ��Ӳ֬��þ�ȶ�FTM�IJⶨû�����Եĸ��š��������������ϵĴ����£��ܹ���ѡ���ԵزⶨFTM�ĺ�����������Ҫ��ƷԤ������
2.5 ��Ʒ�ⶨ��������ʵ��
��2��FMTƬ���㶫�˵�ҩҵ����˾����ʾ��20mg������ȡ20Ƭ�����飬�����ȷ��ȡ0.168 5g FMT����ϡ���������ձ��У���ת��100 mL����ƿ�У����ݣ����ˣ�ȡ��Һ50.0 mL��250 mL����ƿ�У���������ˮ���̶ȣ������Ʒ������Һ��ȷ��ȡһ�������������Ʒ����Һ���ɷݣ���ʵ�鷽��������ƷҺ��FMT�Ķ�����������ip��ֵ����У�������25 mL��ƷҺ��FMT��Ũ�ȣ��������ÿƬҩ����FMT�ĺ�������25 mL��ƷҺ����FMT�Ļ��������飬���ý������1�� ��1 Ƭ����FMT�����IJⶨ��������ʵ��
Tab.1 Determination results of FMT content in tablets and experiment of recovery
�ⶨֵ |
ƽ��ֵ |
RSD |
��ʾ�� |
������ |
����� ��mg�� |
������ Recovery( %) |
19.5 |
19.9 |
1.53% |
20 |
1.69 |
1.68 |
99.41 |
3 ����
3.1 FMT�Ĵ��Ⲩ
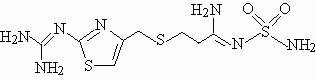
FTM�ṹʽΪ��
Squella��[12]�о�������FMT�ڵι��缫�ϲ��ܻ�ԭ���������Ǵ��Ⲩ����ʵ������У��۲�ڵι��缫���д������ݷų������ú��λ��ⷨ�ڷ��λEp-1.45V�������FMT��Һ1h�������û�����Ե��½�����FMT��ѭ������ͼ��ͼ3,
c������֪���ں�1.0��10-5mol/L FMT��KH2PO4-Na2HPO4 (pH 6.2)������Һ�У���ɨʱFMT������֧�ϲ���һ��ԭ�壬����λEp=
-1.38V��vs SCE��������ɨʱ������֧������Ӧ����������֣��缫��Ӧ���̲����档˵��FMT�������Ǵ��Ⲩ��
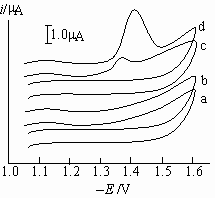
Fig.3 Cyclic voltammetric diagram of FMT
a 0.10mol/L KH2PO4-Na2HPO4 (pH6.2); b a + 2��10-5mol/L K2S2O8;
c a + 1.0��10-5mol/L FMT; d b +1.0��10-5mol/L FMT;
Potential scan rate v = 250 mV/s
���ݴ��Ⲩ������[19]��FMT���Ƚ��ܽ����и����H+���ɾ��е���Ե�FMTH+������FMTH+�ڵ缫������е����ӽ������ɲ��ȶ����м������ɻ�FMTH*�������ɻ�������˫���ӾۺϷ�Ӧ�ų�H2������FMT�Ĵ��Ⲩ������̰�����
FMT + H3+O Û FMTH+ + H2O
FMTH+ + e�C ® FMTH*
2 FMTH* ® 2FMT + H2
3.2 FMT�ĵϣ�ƽ�д��Ⲩ
K2S2O8�����µ�ѭ��������ʵ�飨ͼ3, d����������ɨʱFMT������֧�ϲ���һ��ԭ�壬���λ��δ��K2S2O8��ȣ�������Ep=-1.42V��vs SCE���������ֵ�����˼���10��������ɨʱ������֧��δ���ֻ�ԭ�壬���ǵ��͵ĵ�/ƽ�д��Ⲩ����[18]��
��������K2S2O8����ʱ��S2O82-�������м����FTMH+������������ԭ��S2O82-�Ļ�ԭΪ���������ӵ���������[20]��
S2O82- + e-® SO42- + SO4-·
E ��0.6V��
SO4-· + e-® SO42-������ ����E
��3.4V
S2O82-��ԭ�м�����������ɻ�SO4-���к�ǿ��������������Ҳ�������м���FTMH+��������Ե�FTMH+��S2O82-�����м���FTMH+�ķ�Ӧ�ɱ�ʾ���£�
FMTH* + S2O82-® FMTH++
SO42- + SO4-·
FMTH* + SO4-· ®
FMTH++ SO42-
���ӻ�FTMH+�����ӻ�ԭ���м���FTMH*��S2O82-������FTMH*����FTMH+������γɵ�������ԭѭ����������FTM�ĵ�/ƽ�д��Ⲩ[16]��
[1] Li Xinang, Lu Jinsheng, Huang Hua. Chinese Journal of Pharmaceuticals, 1992, 23: 540-542.
[2] The United States pharmacopeial convention. U.S.P Pharmacopeia's Nation Formulary. XXIII Washington:D.C.January. The board of trustee's official. Revision, 1995: 651��
[3] Pharmacopoeia Committee of PRC. Pharmacopoeia of the People's Republic of China. Part II, 2000Ed,Beijing: Chemical Industry, 2000: 425.
[4] Liu Haitao, Li Li, GuoXiaohong. Pharmaceutical Journal of Chinese People's Liberation Army, 2004, 20: 376-378.
[5] Dowling T C, Frye R F. J Chromatogr B, 1999, 732: 239-244.
[6] Cakir-B, Tosun-Au. Pharm-Sci, 1997, 3/10: 493-495.
[7] Lin Xiaoming, Liu Yanqing. China Pharmacist, 2004, 7: 31-32.
[8] Zhu Min, Song Yi, Zhao Yuxiang. Chinese Journal of Pharmaceuticals, 2002, 33: 348-349.
[9] Chen Weiyang, Hu Yuzhu, Ma Yuefeng, Liu Zhongtao. Chinese Pharmaceutical Journal, 2004, 39: 143-144.
[10] Pan Fuyou. Chinese Journal of Analytical Chemistry, 2001, 29: 243.
[11] J A Squella��C Rivera��I Lemus et al. Mikrochim. Acta. 990, 1: 343-348.
[12] J A Squella��G S Valencia��I Lemus��et al��J-Assoc. Anal Chem.��1989��72: 549-551.
[13] Kang Xiaofeng, Song JunFeng. Science in China (Chemistry), 1999, 42(5): 463-470.
[14] Song JunFeng, Dong Yanhua, Guo Wei, etal. Chinese Journal of Analytical Chemistry, 2001, 29: 745-749.
[15] Kang Xiaofeng, Guo Wei,Zhao Chuan. Science in China (Chemistry), 2000, 43: 275-282.
[16] Guo Wei, Liu Limin, Lin Hong, etal. Science in China (Chemistry), 2002, 45: 158-165.
[17] Liu Limin, Guo Wei, Song JunFeng. Chinese Journal of Analytical Chemistry, 2001, 30: 46-49.
[18] Song J F, Li N, Xu M T. Acta chim sinca, 2004, 62: 78-82.
[19] Mairanovskii S G. J Electroanal Chem, 1963, 6: 77-118.
[20] Memming R J. Electrochem soc, 1969, 116: 785-790.