http://www.chemistrymag.org/cji/2006/084024rc.htm |
Apr. 2, 2006 Vol.8 No.4 P.24 Copyright |
(Anhui Key Laboratory of Functional Molecular Solids, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000)
Abstract Since the discovery and large scale preparation of [60]fullerene, the chemistry of fullerenes has become one of the most developing fields in organic chemistry. Polymeric fullerenes formed by polymerization of the fullerene spheroid possessed unique physical properties, which pioneer one of the new directions for the design of new pattern promising materials based on fullerenes. The progress on the synthesis, magnetic properties and applications of polymeric fullerenes in current years were reviewed.
Keywords Fullerenes, Polymerization, Magnetic property ОлКЯИЛРеЯЉбаОПНјеЙ РющЊЭЅ ЮКЯШЮФ
ЃЈАВЛеЪЁЙІФмадЗжзгЙЬЬхжиЕуЪЕбщЪв АВЛеЪІЗЖДѓбЇЛЏбЇгыВФСЯПЦбЇбЇдК ЮпКў 241000ЃЉ еЊвЊ здДгC60БЛЗЂЯжгШЦфЪЧЗЂеЙГіКъСПжЦБИЗНЗЈвдРДЃЌИЛРеЯЉЛЏбЇвбГЩЮЊгаЛњЛЏбЇбЇПЦЗЂеЙзюПьЕФСьгђжЎвЛЁЃИЛРеЯЉЭЈЙ§ОлКЯЗДгІаЮГЩОпгаЖРЬиЮяРэадФмЕФИЛРеЯЉОлКЯЬхЃЌЮЊЩшМЦКЯГЩаТаЭИЛРеЯЉЛљЙІФмВФСЯПЊБйСЫаТЗНЯђЁЃБОЮФзлЪіСЫОлКЯИЛРеЯЉЕФКЯГЩЗНЗЈЁЂДХадФмМАгІгУНјеЙЁЃ
ЙиМќДЪ ИЛРеЯЉ ОлКЯЗДгІ ДХад
КмОУвдРДЃЌШЫУЧвЛжБШЯЮЊЬМдЊЫижЛгаСНжжЭЌЫивьаЮЬхЃЌМДШсШэЕФЪЏФЋКЭМсгВЕФН№ИеЪЏЃЌЕЋ1985ФъУРЙњПЦбЇМвCurlЁЂSmalleyвдМАгЂЙњПЦбЇМвKrotoвтЭтЕиЗЂЯжСЫЬМдЊЫиЕФЕкШ§жжЭЌЫивьаЮЬх----вдC60КЭC70ЮЊДњБэЕФИЛРеЯЉЁЃздДг1990ФъKraschmerКЭHuffmanЕШВЩгУЪЏФЋАєЕчЛЁеєЗЂЕФЗНЗЈжЦБИКЭЗжРыГіПЫСПМЖЕФC60жЎКѓЃЌC60ЕФИїжжбаОПвдПеЧАЕФЫйЖШЯђЧАЗЂеЙЁЃЦљНёЮЊжЙЃЌC60ЕФбаОПСьгђвбЩцМАЕНгаЛњЛЏбЇЁЂЮоЛњЛЏбЇЁЂЩњУќПЦбЇЁЂВФСЯПЦбЇЁЂИпЗжзгПЦбЇЁЂДпЛЏЛЏбЇЁЂЕчЛЏбЇЁЂГЌЕМЬхЕШжкЖрбЇПЦКЭгІгУбаОПСьгђЃЌВЂдНРДдНЯдЪОГіЦфОоДѓЕФЧБСІКЭживЊЕФбаОПМлжЕЁЃдкжкЖрЕФИЛРеЯЉбмЩњЮяжаЃЌИЛРеЯЉОлКЯЮявбГЩЮЊвЛИіаТЕФбаОПШШЕуЃЌФПЧАгаСНРрОлКЯИЛРеЯЉЃКвЛРрЪЧOЁЂSЁЂSiКЭCЧХСЊНгЕФЖўОлЛђЖрОлИЛРеЯЉ[1]ЃЛСэвЛРрЪЧдкИЛРеЯЉЬМС§жЎМфжБНгППЕЅМќСЌНгЛђ[2+2]ЛЗМгГЩЗДгІаЮГЩЕФОлКЯИЛРеЯЉЁЃКѓвЛРраЭЕФОлКЯИЛРеЯЉОпгавЛаЉВЛбАГЃЕФЬиадЃЌгШЦфЪЧДХадЃЌЪЙЫќНЋРДПЩвдгУгкЙтЕчзгМЦЫуЛњДцДЂКЭМЧТМаХЯЂЕФдЊЦїМўВФСЯЃЌБОЮФзлЪіСЫетРрОлКЯИЛРеЯЉЕФКЯГЩЗНЗЈЁЂадФмМАгІгУНјеЙЁЃ
1 ОлКЯИЛРеЯЉЕФКЯГЩ
ИЛРеЯЉЬМС§жЎМфжБНгЕЅМќСЌНгЛђ[2+2]МгГЩСЌНгаЮГЩОлКЯИЛРеЯЉ,ЦфжЦБИЗНЗЈжївЊгаЛЙдЗЈЁЂИпЮТИпбЙЗЈЁЂЙтгеЕМЗЈЁЂИпЫйеёЖЏФыФЅЗЈЕШЁЃ
1.1 ЙЬЯрЛЙдОлКЯ
C60ЪЧШБЕчзгЯЉЬўЃЌШнвзЛёЕУЕчзгаЮГЩИКРызгЃЌОпгаГЌЕМЕчадЁЂДХадЕШЬиадЁЃ1991ФъHaddonЕШНЋЛЏбЇМЦСПЕФC60КЭН№ЪєМиЕФЛьКЯЮяЗтБедкЪЏгЂУЋЯИЙмРяМгШШЕН260ЁцЃЌ80ЬьКѓвтЭтЕижЦЕУСЫЗлФЉзДЕФОлКЯВњЮя(K1C60)n
[2]ЁЃКѓРДPekker ЕШ[3]дкЙЬЯрЬѕМўЯТЃЌРћгУC60вѕРызгПЩФц[2+2]ЛЗМгГЩЗДгІКЯГЩСЫаБЗНОЇЯЕОлКЯЮя(A1C60)nЃЌЦфжаC60вѕРызгЪЧЯпаЭОлКЯЮя(ЭМ1)ЃЌвбБЛXRDЪ§ОнжЄЪЕЁЃбаОПЗЂЯжЃЌетбљЕФИЛРеЯЉЧХСЊХХСаНсЙЙФмЙЛБэЯжГіГпДчЩЯЕФЖрбљадКЭСюШЫИааЫШЄЕФРрЫЦгкН№ЪєадЕФЕчбЇКЭЪвЮТЬњДХадЕФЬиадЁЃе§вђЮЊШчДЫЃЌ(A1C60)n
(A=K, Rb КЭCs)РраЭЕФЛЏКЯЮяГЩЮЊСЫбаОПЕФШШЕуЁЃР§ШчЃЌЕМЕчОлКЯЮя(K1C60)nПЩХрбјГЩЪЎЗжжЎМИКСУзГЄЕФЕЅОЇЃЌЦфОлКЯЖШГЌЙ§СЫ100,000[4]ЁЃЯждквВПЩвдЭЈЙ§ЙВеєЗЂЗНЗЈжЦБИ(K1C60)nЕЅОЇЃКМДАбЛЏбЇМЦСПЕФC60КЭKЗтзАдкЪЏгЂЙмРяЃЌдкЮТЖШЬнЖШБфЛЏЕФТЏзгРяМгШШЃЌC60КЭKЗжБ№БЛМгШШЕН600ЁцКЭ150ЁцЃЌОЙ§МИИіаЁЪБЕиМгШШЃЌдкТЏжаДѓдМ300ЁцЕФЧјгђЃЌгаK1C60ЕФЮЂОЇБЁФЄИВЕНСЫЙмБкЩЯЁЃдкЮЂОЇЛљжЪжаЯтЧЖЕФ(K1C60)n
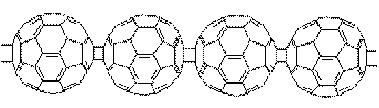
ЭМ1 (C60)nОлвѕРызгЪОвтЭМ
Fig. 1 Schematic depiction of the C60 polyanions
1.2
ИпЮТИпбЙОлКЯИпЮТИпбЙЪЧгУгкC60ОлКЯЗДгІзюЯШНјЕФММЪѕЁЃЗлФЉзДC60ПЩдкИпЮТИпбЙЯТзЊБфЮЊСНжжЯрЖдВЛЮШЖЈЕФЮяЯр[5]ЁЃЕквЛжжЪЧдк5GPaЕФбЙСІЯТНЋC60МгШШЕН300Ёц-400ЁцВњЩњЕФОЇИёВЮЪ§ЮЊ13.6ÅЕФУцаФСЂЗНfccаЭC60ОлКЯЮяЃЛСэвЛжждђЪЧдкЭЌбЙЯТНЋC60МгШШЕН500Ёц-800ЁцКѓаЮГЩЕФОЇИёВЮЪ§ЮЊa=9.22ÅЃЌc=24.6ÅЕФаБЗНСљУцЬхЃЈRЃЉаЭC60ОлКЯЮяЁЃСНжжОлКЯЮядкгаЛњШмМСжаЕФШмНтЖШКмЕЭЃЌЫЕУїдкИпЮТИпбЙЙ§ГЬжаC60ЗжзгМфЕФОрРызуЙЛаЁвджТгкПЩвдаЮГЩЛЏбЇМќЁЃСэЭтЃЌКьЭтЁЂРТќКЭКЫДХЙВеёбаОПБэУїЃЌдкОлКЯЙ§ГЬжаC60ЖўЪЎУцЬхЖдГЦадМБОчНЕЕЭЁЃВюЪОЩЈУшСПШШЗЈКЭКьЭтЙтЦзВтЪдЗЈвбБЛгУгкбаОПC60ЕФСНжжбЙСІОлКЯЮяЯрЕФНтОлЙ§ГЬЃЌдкP=1atmЯТ, аБЗНСљУцЬх(R)КЭУцаФСЂЗН(fcc)C60ОлКЯЮяПЩзЊБфЮЊЗлФЉзДЕФC60ЁЃгаШЄЕФЪЧбЙСІгеЕМЕФC70ОлКЯжЛаЮГЩЕЅвЛЕФ[2+2]МгГЩВњЮяЖўОлЬхC140[6]ЁЃ
C60дкОЇЬхжаППЗЖЕТЛЊСІОлМЏЃЌОЙ§ИпбЙКЭИпЮТДІРэЃЌC60ЗжзгМфЭЈЙ§ЙВМлМќНсКЯзЊБфЮЊОлКЯЯр [7,8]ЁЃC60ЗжзгМфМќЕФЪ§СПКЭОлКЯЮяЕФНсЙЙвРРЕгкОлКЯЬѕМўЃЌИФБфОлКЯЬѕМўПЩвдКЯГЩОпгаИїжжИїбљЮяРэЬиадЕФвЛЮЌ(е§НЛОЇЯЕ)ЁЂЖўЮЌ(аБЗНСљУцЬхКЭЫФУцЬх) ЕФОлКЯЮяЁЃФПЧА,ИпЮТИпбЙжЦБИЕФC60ОлКЯЮяжавбгаЫФИіОлКЯЮяЯрЃЌМДЖўОлЬх(D)ЁЂе§НЛОЇЯЕ(O)ЁЂЫФУцЬх(T)КЭаБЗНСљУцЬх(R)ЁЃDЯргЩC60ЖўОлЬхзщГЩЃЌOЯрЪЧвЛИівЛЮЌ(1D)ЯпаЭОлКЯЮяЃЌЖјTЯрКЭRЯрИїздДњБэзХВЛЭЌЕФЫФУцЬхКЭаБЗНСљУцЬхЖўЮЌ(2D)C60ОлКЯЮя(ЭМ2)ЁЃМђЕЅЕиЫЕЃЌВњТЪзюИп(70%)ЕФDЯрЪЧдкP=1.5GPaЃЌT=423KЕФЬѕМўЯТЗДгІ1000sаЮГЩЕФЃЛOЯрЪЧдкP=1.2GPaЃЌTЃН570KЕФЬѕМўЯТЗДгІ20000sЩњГЩЕФЃЛTЯрЪЧдкP=2.2GPaЃЌTЃН873KЕФЬѕМўЯТЗДгІ2000sЩњГЩЕФЃЛRЯрЪЧдкP=6.0GPaЃЌTЃН873KЕФЬѕМўЯТЗДгІ2000sаЮГЩЕФЃЌДЫЭтЃЌдкPЃН6.0GpaЃЌTЃН1025KЁЋ1050KЕФЬѕМўЯТЩњГЩЕФвЛВПЗжRЯрбљЦЗБЛГЦзїДХадЬМЁЃбаОПжЄЪЕЃЌФПЧАЕЅЖРЪЙгУКьЭтКЭРТќЙтЦзПЩвдКмКУЕиЖЈСПЪЖБ№етаЉОлКЯЯр[9]ЁЃ
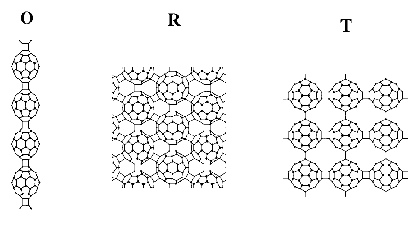
ЭМ2 вЛЮЌКЭЖўЮЌC60ОлКЯЮяЕФНсЙЙЪОвтЭМ
Fig.2 Schematic structures of 1D and 2D C60 polymers
1.3 ЙЬЬЌдЄзщзАОлКЯ
C60ОпгаШ§ЮЌХХСаЕФЗДгІЛюадЫЋМќЃЌетОЭдьГЩСЫКмФбПижЦЖрМгГЩЗДгІВњЮяЕФНсЙЙЁЃЙЬЬЌдЄзщзАЬсЙЉСЫвЛИігааЇЕФНтОіЗНЗЈЃЌМДЭЈЙ§C60КЭЦфЫќЗжзгЙВНсОЇЃЌНЋC60ЗтзАдкЖРСЂЕФЕЅдЊЃЌC60ЗжзггаађХХСа,ЪЙC60ЗжзгМфжЛФмдкЬиЖЈЕФЗНЯђНјааОлКЯЁЃШчдкГЌЗжзгЙВНсОЇЬхC60(ET)2(ET=Жў(ввЯЉЖўСђЛљ)ЫФСђИЛЭпЯЉ)жаЃЌИпЮТИпбЙФмИќгааЇЕигеЕМC60ЖўОлГЩC120[10]ЁЃC60гыБЗМЬўЙВНсОЇПЩвдЪЙC60аЮГЩбЯИёЕФжБЯпХХСа(ЭМ3)ЁЃБЗМЬўзшжЙСЫC60НЛВцСЊНгЃЌИпЮТИпбЙЯТЛёЕУСЫЯпаЭC60ОлКЯЮя[11]ЁЃдЄзщзАЕФC60гаРћгкЗжзгМфНјаа[2+2]ЛЗМгГЩЗДгІ[12]ЁЃ
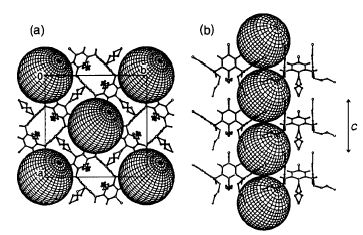
ЭМ3 C60КЭБЗМЬўЕФОЇЬхЖбЛ§ЪОвтЭМ:ЃЈaЃЉИЉЪг
ЃЈbЃЉВрЪг
Fig.3 Two views of the crystal packing of C60
and calixarene molecules (a) looking down the columns and (b)side-on.
1.4 ЙтгеЕМОлКЯ
C60дкЙЬЯржавзЗЂЩњИпаЇЕФЙтОлКЯЗДгІЃЌМД[2+2]ЙтМгГЩЗДгІЁЃЙтгеЕМИЛРеЯЉЖўОлЗДгІ(ЛђОлКЯЗДгІ)ФПЧАБЛЯожЦдкЙЬЬхбљЦЗ[13,14]КЭаќИЁвК[15]ЬхЯЕжаЁЃЭЈЙ§ГЌЗжзгздзщзАЃЌВЂСаЗДгІЫЋМќ,ПЩвдМгЫйЙтЗДгІВњЮяЕФаЮГЩЁЃC60бмЩњЮяЭЈЙ§ЛЅВЙЕФЧтМќНсКЯЃЌНЋЕЅИіC60бмЩњЮяЗжзгАДеевЛЖЈЕФЫГађзщзА(ЭМ4),
дйНјааЙтОлКЯдђаЮГЩЖўОлЬх[16]ЁЃГЌЗжзгЬхЯЕжаЗжзгМфОпгаКмИпЕФНсКЯГЃЪ§ЃЌПЩвдБЛгУгкЙВОлЮяКЭЭЌОлЮяЕФаЮГЩ,ВЂЕУЕНИќЖрбљЕФНсЙЙЁЃ
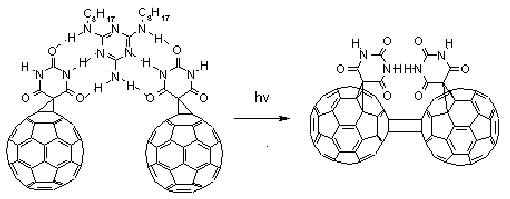
ЭМ4 ДцдкЛЅВЙЧтМќЕФC60бмЩњЮяЗЂЩњЙтгеЕМОлКЯЪОвтЭМ
Fig.4 Schematic depiction of the
photodimerization of C60 derivatives in the presence of complementary
hydrogen-bonding
1.5 ЯогђМгШШОлКЯ
дкЮЂЗДгІЦїжаНјааОлКЯЗДгІ,РћгУЯогђаЇгІПЩПижЦаЮГЩЕФОлКЯЮяЕФНсЙЙЁЃC60OдкЕЅБкЬМФЩУзЙмжаБЛМгШШКѓаЮГЩСЫЯпзДОлКЯЮя[17](ЭМ5), гыжБНгМгШШC60OаЮГЩЕФжІзДШ§ЮЌНсЙЙЕФОлКЯЮяВЛЭЌЁЃРћгУвЛЮЌЯогђаЇгІВЛЪЇЮЊКЯГЩЯпаЮОлКЯЮяЕФКУЗНЗЈЁЃ
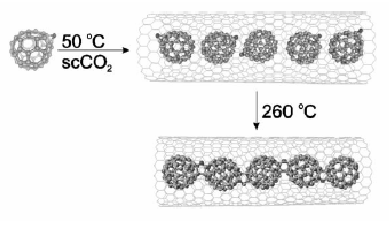
ЭМ5 C60OдкЕЅБкЬМФЩУзЙмжаЗЂЩњЛЏбЇЗЂгІЕФЪОвтЭМ
Fig.5 Schematic depiction of the
polymerization of C60O inside single-walled carbon nanotubes
1.6 вКЯрЛЙдОлКЯ
C60гыCp*2CrЗжБ№дкСкЖўТШБНКЭБНжаЗДгІЃЌПЩаЮГЩХфКЯЮяCp*2Cr·C60·(C6H4Cl2)2КЭCp*2Cr·C60·(C6H6)2ЁЃЕЅОЇНсЙЙВтЖЈБэУїCp*2Cr·C60·(C6H4Cl2)2ОЇЬхжаДцдкЖўОлЮя(C60)2ЈC[18](ЭМ6)ЁЃНќКьЭтЙтЦзБэУї, C60дквКЯржаБЛCp*2CrЛЙдЮЊC60·-
[19]ЃЌПьВНжшаЮГЩСЫCp*2CrЃЋКЭC60·-РызгЃЛЕкЖўВНЪЧТ§ВНжшЃЌаЮГЩСЫЙВМлСЊНгЕФCp*2Cr+- C60·-ЃЌНєНгзХЗЂЩњздгЩЛљЕФдйНсКЯЃЌзюжеаЮГЩСЫCp*2Cr-C60РраЭЕФХфКЯЮяЁЃЭЌбљЃЌдкCTV·(Cs)2·(C70)2·(DMF)7·(C6H6)0.75[20]ЁЂCr(C6H6)2·C70·C6H5Me[21]КЭTDAE·C70·C6H5Me[22]ЕЅОЇжавВЗЂЯжСЫЕЅМќСЌНгЕФ(C70)2ЈCЖўОлЮя(ЭМ7)ЁЃ
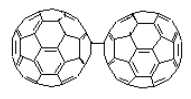
ЭМ6 Cp*2Cr·C60·(C6H4Cl2)2ОЇЬхжа(C60)2ЈCЖўОлЮяЕФЗжзгНсЙЙЪОвтЭМ
Fig.6 (C60)2ЈC
molecule structure in Cp*2Cr·C60·(C6H4Cl2)2
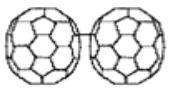
ЭМ7 CTV·(Cs)2· (C70)2·(DMF)7·(C6H6)0.75жа(C70)2ЈCЖўОлЮяЕФЗжзгНсЙЙЪОвтЭМ
Fig.7 (C70)2- structure in CTV·(Cs)2·
(C70)2·(DMF)7·(C6H6)0.75
гаШЄЕФЪЧ
Jansen СьЕМЕФаЁзщЭЈЙ§дквКАБжаЪЙгУН№ЪєБЕЛЙдC70ЕквЛДЮКЯГЩСЫЯпаЭОлКЯЕФ(C702-)nРызг[23]ЃЌЫќДцдкгкБЕбЮ[Ba(NH3)9]C70·7NH3жаЃЌБЛЕЅОЇXЩфЯпЗжЮіжЄЪЕ(ЭМ8)ЁЃЭЈЙ§СНИіДІгкЯрЗДЮЛжУЮхдЊЛЗЩЯЕФЬМдзгЕФГЩМќзїгУЃЌC70С§ЯпаЭСЊНгЃЌаЮГЩСЫдк[001]ЦНУцФк(C702-)nЕФЧсЮЂОтГнаЮХХСа,ЧвЧХСЊЬМдзгЪЧsp3дгЛЏЁЃ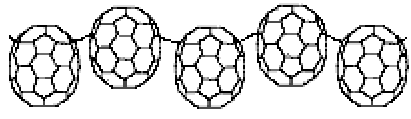
ЭМ8 (C702-)nРызгЕФНсЙЙЪОвтЭМ
Fig.8 Schematic depiction of the structure of (C702-)n
1.7 ИпЫйеёЖЏФыФЅЗЈОлКЯ
ЭѕЙйЮф[26]ЕШРћгУИпЫйеёЖЏФыФЅЗЈ(HSVM)ЃЌдкЙЬЯрЬѕМўЯТНЋC60гыKCNЗДгІЃЌКЯГЩСЫC60ЖўОлЮяC120ЁЃИУЗДгІгаСНжжПЩФмЛњРэЃЌвЛЪЧЧзКЫЛњРэЃЌСэвЛЪЧШчЭМ9ЫљЪОЕФздгЩЛљЛњРэЁЃздгЩЛљЛњРэПЩФмИќЮЊКЯРэЃЌвђЮЊЩйСПЕФЛЙдадН№ЪєПЩвдгааЇЕив§ЗЂСДдіГЄЙ§ГЬЁЃ
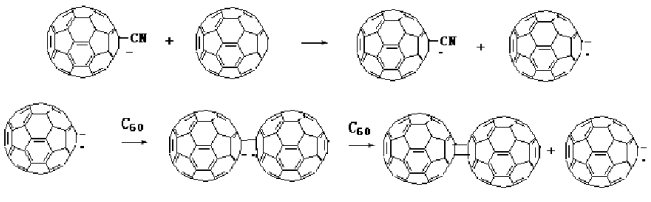
ЭМ9 здгЩЛљЛњРэЪОвтЭМ
дкМибЮ(K2CO3,CH3CO2K)ЛђЛЙдадН№Ъє(Li, Na, K, Mg, Al, Zn)ЛђгаЛњМюДцдкЕФЬѕМўЯТЃЌЭЈЙ§HSVMЗЈвВПЩвджЦБИГіC120[27],ЭЌЗЈЛЙПЩжЦБИC70ЖўОлаЮГЩC140ЕФЮхжжЭЌЗжвьЙЙЬх[28]ЁЂC60КЭC70НЛВцЖўОлЮяC130[29]ЁЂC60Ш§ОлЮяC180ЕФИїжжЭЌЗжвьЙЙЬх[30]ЁЃC180ЕФГЩМќЬиеїКЭЭЌЗжвьЙЙЬхЕФНсЙЙПЩгУЫэЕРЩЈУшЕчОЕ(STM)Бэеї[31]ЁЃ
2ЃЎДХадФмИЛРеЯЉЗжзгМфМќКЯЕФЪ§СПКЭОлКЯЮяЕФНсЙЙВЛЭЌ,ЕМжТОлКЯИЛРеЯЉДцдкгаађЛђЮоађзДЬЌ,ОпгаН№ЪєКЭ/ЛђАыЕМЬхЕФЬиадЕШЁЃ
C60ЪЧПЙДХадЕФЃЌЖјЛљгкC60ЕФвЛаЉВФСЯЪЧЬњДХадЕФЁЃвбБЈЕРЕФИЛРеЯЉЛљЗжзгЬњДХЬхжївЊгаСНРрЃКЕквЛРрЪЧИЛРеЯЉЕчКЩзЊвЦбЮ,ШчTDAE- C60ЃЈTDAE =ЫФЃЈЖўМзЛљАБЛљЃЉввЯЉЃЉЃЌTc=16K[32]ЃЛ[3-АБЛљБНЛљМзГХC60][Cp2Co]ЃЌTc=19K[33]. КЭTTFxC60Bry[34]ЁЃЕкЖўРрЬњДХЬхЪЧC60ЖўЮЌВузДОлКЯЮяЁЃзюНќЕФЪЕбщКЭРэТлбаОПБэУїгЩгкДПЪЏФЋжаЕчзгЕФВЛЮШЖЈадЃЌПЩвдв§Ц№ЪЏФЋВњЩњЪвЮТГЌЕМКЭЬњДХаджЪЁЃетвЛНсЙћв§Ц№СЫШЫУЧбаОПДПЬМЙЬЬхЕФЙуЗКаЫШЄЁЃ2001ФъMakarovaЕШШЫдкЬНбАC60ОлКЯЮяЕФГЌЕМЯжЯѓЪБвтЭтЕиЗЂЯжСЫC60ЖўЮЌВузДОлКЯЮяСтУцОЇЬхОпгаЬњДХадНЛЛЛЯрЛЅзїгУЃЌTcИпДя500K[35, 36]ЃЌетЪЧЦљНёЮЊжЙTcзюИпЕФДПгаЛњЬњДХЬхЃЌЪЧЗжзгЛљЬњДХЬхбаОПСьгђЕФвЛИіжиДѓЭЛЦЦЃЌОпгаживЊЕФбЇЪѕвтвхЁЃСэЭт,ЙтбѕЛЏC60КѓTCПЩДя800K[37]ЁЃEu9C70 Tc=38K [38]ЁЃЧтЛЏИЛРеЯЉC60H24жаЗЂЯжгаЬњДХад[39]ЃЌЦфTCжЕДѓгк300KЁЃгаШЄЕФЪЧЃЌНЋC60H24ДЂВивЛФъКѓЃЌзюГѕЕФЬњДХадзЊБфЮЊПЙДХадЃЌетвЛНсЙћЫЕУїC60H24ЕФЬњДХадВЛПЩФмРДздДХаддгжЪЁЃ
ИЛРеЯЉЛљДХЬхгаМИИіжївЊЕФЬиеїЃК(1) дкИЛРеЯЉЕЅдЊжЎМфЕФ p Ечзгзда§ёюКЯПЩвдВњЩњЬњДХадЃЌЖјВєдгМСВЛЖдзда§гаађзїГіЙБЯзЃЛ(2) жЦБИЙ§ГЬКЭДХадЬиеїжЎМфДцдкКмЧПЕФЯрЛЅЙиЯЕЁЃВЛЬЋДѓЕФБЅКЭДХЛЏЧПЖШКЭДХадЖдгкЮТЖШЗЧе§ГЃЕФвРРЕЙиЯЕЃЌКмПЩФмЪЧгЩгкбљЦЗЕФФЩУзОЇЬхБОадОіЖЈЕФЃЛ(3) ЛёЕУЬњДХадЮяЯрЕФЙиМќЪЧСкНќИЛРеЯЉЗжзгЯрЛЅШЁЯђ[38,40]ЁЃC60вѕРызгЕФЯрЛЅШЁЯђОіЖЈСЫЗжзгМфДХНЛЛЛЙиЯЕЁЃ
ИЛРеЯЉЛљЬњДХЬхЕФЬњДХаХКХЪЧгЩИЛРеЯЉв§Ц№ЕФ,двђгаЃК(1) ЫцЛњЗжВМЕФдгжЪЬњРызгВЛФмВњЩњЬњДХадЃЌвђЮЊдзгжЎМфЕФОрРыЬЋДѓЃЌДЋЕМЕчзгЕФУмЖШЬЋЕЭЃЛ(2) МДЪЙЫљгаЕФЬњдзгаЮГЩСЫКъЙлДиШКЃЌВтЕУЕФC60ДХЛЏЧПЖШдкФГаЉЧщПіЯТГЌЙ§СЫЙРЫудгжЪЙБЯзЕУЕНЕФжЕЃЛ(3) бљЦЗЕФДХадЪмбЙСІКЭЮТЖШЕШОлКЯЬѕМўПижЦЃЛ(4) дгжЪВЛФмНтЪЭдкC60ЙтОлКЯЙ§ГЬжаЬњДХаХКХЕФМгЧП[37, 41]ЃЛ(5) ЖдгкдкдгжЪХЈЖШаЁгк1 ppmЕФКъЙлЧјгђЕФДХГыНсЙЙбаОПБэУїЃЌИЛРеЯЉЕФЬњДХадЪЧЦфЙЬгаЪєадЁЃ
ОлКЯИЛРеЯЉЬњДХадЕФЛњРэПЩФмЪЧгЩгкC60РызгЕФОжВПДХОиКЭСїЖЏЕчзгжЎМфЕФёюКЯ[42]Лђsp3ЬМдзгЗжПЊЕФСНИіsp2ЬМздгЩЛљжЎМфЕФЬњДХЯрЛЅзїгУ[43]ЛђдкНсЙЙШБЯнКЭsp3ЬМжЎМфДцдкЯрЛЅзїгУ[44]ЁЃИЛРеЯЉОлКЯЙ§ГЬжаВњЩњЕФАќКЌsp2КЭsp3дгЛЏЬМдзгЕФНсЙЙЃЌжЇГжC60ЕФЬњДХадЁЃдкИпбЙгеЕМИЛРеЯЉОлКЯЕФЙ§ГЬжаЃЌДХадГіЯждкИЛРеЯЉС§НЋвЊЦЦЫщЕФЧщПіЯТ,ЦЦЫщЕФИЛРеЯЉЗжзгМфГЩМќЛђС§ЕФШБЫ№дьГЩСЫВЛГЩЖдзда§ЁЃЗжзгбѕЙтМЄЗЂКѓЗжНтЃЌгыИЛРеЯЉЯрЛЅзїгУВњЩњОжВПзда§ЁЃдкЮобѕЕФЬѕМўЯТЃЌЙтбѕЛЏВЛФмИФБфИЛРеЯЉЕФДХадЁЃЖдгкОлКЯИЛРеЯЉЬњДХадааЮЊЕФеце§Ц№вђФПЧАЩаВЛФмШЗЖЈЃЌгаД§гкНјвЛВНбаОПЁЃ
3 еЙЭћ
ФПЧАЖдгкОлКЯИЛРеЯЉЕФбаОПЛЙКмЩйЃЌКЯГЩЗНЗЈжЛгаИпЮТИпбЙЗДгІНЯЮЊГЩЪьЃЌЦфЫќЕФЗНЗЈвВжЛЪЧИеИеЦ№ВНЃЌЛЙдкбаОПКЭЬНЫїНзЖЮЃЛВњЮявдОлКЯC60ОгЖр,ЦфЫќЕФИЛРеЯЉОлКЯЛЙЩйМћБЈЕРЃЛШЫУЧЖдОлКЯИЛРеЯЉЕФаджЪСЫНтвВжЛЭЃСєдкБэУцЯжЯѓНзЖЮЁЃC60ЪЧ20ЪРМЭвЛИіеёЗмШЫаФЕФЗЂЯжЃЌЫќЮЊЛЏбЇбЇПЦПЊБйСЫвЛИіеИаТЕФбаОПСьгђЃЌЖјОлКЯИЛРеЯЉдђЪЧвЛеХИеаДСЫМИБЪЕФАзжНЃЌвЛЖЈгаОоДѓЕФЗЂеЙПеМфКЭЧБдкЕФгІгУМлжЕЁЃЮвУЧШЯЮЊОлКЯИЛРеЯЉЕФбаОПШШЕуКЭЗЂеЙЗНЯђжївЊЬхЯждквдЯТМИИіЗНУцЃК(1)
ЬНЫїЮТКЭЬѕМўЯТКъСПжЦБИОлКЯИЛРеЯЉЕФаТЗНЗЈЃЛ(2)
ЬНЫїКЯГЩИЛРеЯЉбмЩњЮяОлКЯЮя,
баОПМгКЯЛљЭХЖдОлКЯИЛРеЯЉбмЩњЮяадФмЕФгАЯь,ЩшМЦКЯГЩаТгБЕФИЛРеЯЉОлКЯЮяЬњДХЬх;
(3)
ЯЕЭГЁЂЩюШыЕибаОПОлКЯИЛРеЯЉНсЙЙКЭадФмЃЌЗЂЯжЙцТЩЃЌЬНОПБОжЪЁЃЯраХЫцзХЖдОлКЯИЛРеЯЉбаОПЕФНјвЛВНЩюШыЃЌЦфБОжЪКЭгУЭОвВЛсдНРДдНЖрЕиБЛШЫУЧШЯЪЖЁЂРэНтКЭРћгУЁЃ
[1] Yin J J, Li Y G, Li B. et al. Chem. Commun., 2005: 3041-3043 and references therein.
[2] Haddon R C, Hebard A F, Rosseinsky M J. et al. Nature, 1991, 350: 320-322.
[3] Pekker S, Forró L, Mihály L. et al. Solid State Commun., 1994, 90: 349-352.
[4] Pekker S, Jánossy A, Mihaly L. et al. Science, 1994, 265: 1077-1078.
[5] Iwasa Y, Arima T, Fleming R M. et al. Science, 1994, 264: 1570-1572.
[6] Lebedkin S, Hull W E, Soldatov A. et al. J. Phys. Chem.B 2000, 104: 4101-4110.
[7] Palacio F. Nature, 2001, 413: 690-691.
[8] Korobov M V, Senyavin V M, Bogachev A G. et al. Chem. Phys. Lett., 2003, 381: 410-415.
[9] Senyavin V M, Davydov V A, Kashevarova L S. et al. Chem. Phys. Lett., 1999, 313: 421-425.
[10] Iwasa Y, Tanoue K, Mitani T. et al. Chem. Commun., 1998, 13: 1411-1412.
[11] Barbour L J, Atwood J L. Chem. Commun., 1998, 17: 1901-1902.
[12] Knol J, Hummelen J C. J. Am. Chem. Soc., 2000, 122: 3226-3227.
[13] Rao A M, Zhou P, Wang K A. et al. Science, 1993, 259: 955-957.
[14] Zhou P, Dong Z H, Rao A M. et al. Chem. Phys. Lett., 1993, 211: 337-340.
[15] Sun Y P, Ma B, Bunker C E. et al. J. Am. Chem. Soc., 1995, 117: 12705-12711.
[16] McClenaghan N D, Absalon C, Bassani D M. J. Am. Chem. Soc., 2003, 125: 13004-13005.
[17] Britz D A, Khlobystov A N, Porfyrakis K. et al. Chem. Commun., 2005: 37-39.
[18] Konarev D V, Khasanov S S, Otsuka A. et al. J. Am. Chem. Soc., 2002, 124: 8520-8521.
[19] Konarev D V, Drichko N V, Graja A. J. Chem. Phys., 1998, 95: 2143-2156.
[20] Konarev D V, Khasanov S S, Vorontsov I I. et al. Chem. Commun., 2002: 2548-2549.
[21] Yoshida Y, Otsuka A, Konarev D V. et al. Synth. Met., 2003, 133-134: 703-705.
[22] Oshima K, Kambe T, Fujiwara M. et al. Synth. Met., 2003, 133-134: 609-701.
[23] Brumm H, Peters E, Jansen M. Angew. Chem. Int. Ed., 2001, 40: 2069-2071.
[26] Wang G W, Komatsu K, Murata Y. et al. Nature, 1997, 387: 583-586.
[27] Komatsu K, Wang G W, Murata Y. et al. J. Org. Chem., 1998, 63: 9358-9366.
[28] Forman G S, Tagmatarchis N, Shinohara H. J. Am. Chem. Soc., 2002, 124: 178-179.
[29] Komatsu K, Fujiwara K, Murata Y. Chem. Commun., 2000, (17): 1583-1584.
[30] Komatsu K, Fujiwara K, Murata Y. Chem. Lett., 2000: 1016-1017.
[31] Kunitake M, Uemura S, Ito O. et al. Angew. Chem. Int. Ed., 2002, 41: 969-972.
[32] Allemand P M, Khemani K C, Koch A. et al. Science, 1991, 253: 301-303.
[33] Mrzel A, Omerzu A, Umek P. et al. Chem. Phys. Lett. 1998, 298:329-334.
[34] Li Y L, Zhang D Q, Bai F L. et al. Solid State Commun. 1993, 86: 475-477.
[35] Makarova T L, Sundqvist B, H?hne R. et al. Nature, 2001, 413: 716-718.
[36] H?hne R, Esquinazi P. Adv. Mater. 2002, 14:753-756.
[37] Murakami Y, Suematsu H. Pure Appl. Chem., 1996, 68: 1463-1467.
[38] Takenobu T, Chi D H, Margadonna S. et al. J. Am. Chem. Soc., 2003, 125: 1897-1904.
[39] Antonov V E, Bashkin I O, Khasanov S S. et al. J. Alloys Compd., 2002, 330: 365-368.
[40] Narymbetov B, Omerzu A, Kabanov V V. et al. Nature, 2000, 407: 883-885.
[41] Makarova T L, Han K ЈCH, Esquinazi P. et al. Carbon, 2003, 41: 1575-1584.
[42] Jarlborg T. Phys. Rev. Lett., 2000, 85: 186-189.
[43] Coey J M D, Venkatesan M, Fitzgerald C B. et al. Nature, 2002, 420: 156-159.
[44] Andriotis A N, Menon M, Sheetz R M. et al. Phys. Rev. Lett., 2003, 90: 026801-1-026801-4.