http://www.chemistrymag.org/cji/2006/084025prc.htm |
Apr. 2, 2006 Vol.8 No.4 P.25 Copyright |
Separation and determination of seven fluoroquinolones in pork sample by HPLC-PAD
Liu Pengyan,Wang Hongyu,Jiang Ning,Tan Zhiqiang
(College of Chemistry and Environmental Science, Hebei University, Baoding, 071002)
Keywords HPLC; PAD; fluoroquinolones; SPE; pork
HPLC-PAD法同时测定猪肉中7种氟喹诺酮类兽药的残留
刘芃岩 王洪宇 姜宁 谭志强(河北大学化学与环境科学学院,河北保定 071002) 2006年1月9日收稿;河北省自然科学基金项目(B2005000105)
摘要 建立了一种二极管阵列-高效液相色谱同时分离测定猪肉中7种氟喹诺酮类兽药残留的方法(恩诺沙星、环丙沙星、左氧氟沙星、依诺沙星、诺氟沙星、氟罗沙星及洛美沙星)。样品采用乙腈/醋酸提取,经C18 固相萃取小柱净化后,进HPLC系统检测。色谱柱为Shimadzu VP-ODS(250 mm×4.6 mm, 5
mm);流动相为甲醇-0.02 mol/L磷酸盐缓冲液-三乙胺系(pH 2.8);检测波长281 nm。检出限达到9~14 mg/kg;标准曲线在0.05~5mg/mL范围内线性关系良好(R>0.999 9);加标回收率除依诺沙星在66%~71%外,其它6种均在80%~92%,RSD<5% (n=3)。该方法准确、灵敏、可操作性强。关键词 高效液相色谱法;二极管阵列检测器;氟喹诺酮;固相萃取;猪肉 1 引言
氟喹诺酮(fluoroquinolones, FQs)类药物,具有抗菌谱广、高效、低毒、组织穿透力强和价格低廉的特点,故在医学和兽医学中应用广泛。但其残留可导致人体内病原体的耐药性[1,2],而且氟喹诺酮类药物对儿童中枢神经系统的副作用也有报道[3],所以欧盟(EU)和FAO/WHO食品添加剂与污染物联合专家委员会(JECFA)早在1990年就规定了几种喹诺酮的最大残留量[4]。目前关于氟喹诺酮类残留分析方法的报道较多,通常采用液相色谱法结合荧光检测器[5-11]、紫外检测器[12-14]、质谱检测器[15-17]分离测定或采用毛细管电泳[18,19]。而采用高效液相色谱-二极管阵列检测器法分离检测多种FQs的文献较少[20],在此文献中报道了动物组织中诺氟沙星、麻保沙星、恩诺沙星、环丙沙星和双氟沙星等5种FQs的分离检测,回收率除了诺氟沙星在98%以上外,其它几种的回收率较低(70%-82%);本文采用猪肉作样品,经过适当的前处理,7种FQs用高效液相色谱-二极管阵列检测法(HPLC-PAD)得到了很好的分离,且方法的检出限和回收率均达到了残留分析的要求。本文采用HPLC-PAD对FQs分析,实现了紫外吸收在线图谱扫描,极大的方便了检测物质的定性和检测波长的选择,二极管阵列检测器能从保留时间和紫外光谱两方面对FQs进行定性,使结果更准确。 2 实验部分
2.1 主要仪器与试剂
2.1.1. 仪器 日本岛津LC-10Avp高效液相色谱仪,配SPD-M10Avp检测器和SIL-HTA自动进样器;FSH-2型匀浆机(江苏仪通电子有限公司);C18 SPE小柱(500 mg/3 mL,Spe-edTM,Applied separations Inc.);LD-4型离心机(江苏亿通电子有限公司),SK-1型快速混匀器(常州国华电器有限公司)。
2.1.2. 试剂 氟罗沙星(Fleroxacin, FLX),诺氟沙星(Norfloxacin, NOR),环丙沙星(Ciprofloxacin, CIP),依诺沙星(Enoxacin, ENO),左氧氟沙星(Levofloxacin, LVFX),洛美沙星(Lomfloxacin, LOM)(中国药品生物制品检定所),恩诺沙星(Enrofloxacin, ENR)(德国Dr. Ehrenstorfer公司),甲醇、乙腈为色谱纯,其它试剂均为分析纯,水为二次蒸馏水。
2.2 色谱条件
色谱柱:Shimadzu VP-ODS(250 mm×4.6 mm, 5 mm);流动相:甲醇-0.02 mol/L磷酸盐缓冲液/三乙胺系(25:75),流速:1.0 mL/min;柱温:40℃;进样量10 mL。此条件下,7种FQs能得到有效的分离,如图1(a)所示。
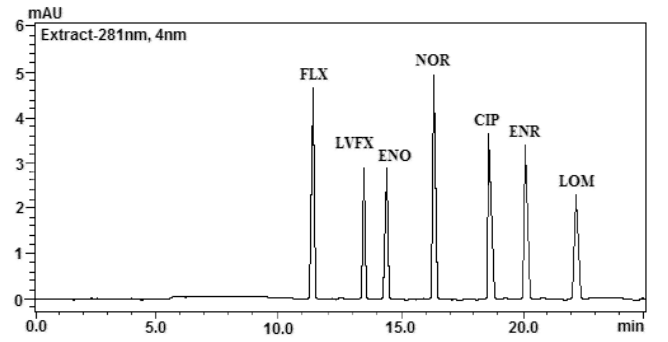
图1 7种氟喹诺酮类药物的色谱图
Fig.1 Chromatogram of seven fluoroquinolones 2.3 标准溶液的配制
2.3.1 储备液 准确称取7种FQs标准品各4.00 mg,分别于7个10 mL容量瓶中,用甲醇溶解并定容,作为单标储备液,-18℃保存。
2.3.2 工作液 准确移取各单标储备液适量于25 mL容量瓶中,配制成浓度为10 mg/mL的标准混合工作溶液;4℃下保存,保存期1个月。
2.4 样品处理
2.4.1 空白样品 将未检测到氟喹诺酮类药物的新鲜的肉样切碎,每份5.0 g分装在离心管中,在< -18℃条件下冷冻备用,制成空白样品。
2.4.2 加标样品 空白样品在常温下解冻后,向其中加入一定量的标准混合溶液,暗处静置2~3小时。
2.4.3 样品提取 在空白和加标样品中,均加入一定量的标准混合溶液和8.0 mL 1%的醋酸/乙腈提取液,匀浆后以4 000 rpm离心10 min,转移出上清液,样品中再加入8.0 mL提取液重复以上操作,残渣及匀浆杯、刀用4.0 mL提取液洗涤后离心,合并3次上清液,再离心1次,取上清液10.0 mL用正己烷萃取3次(3、2、2 mL),涡旋混匀,弃去上层,下层溶液在50±2℃下,氮吹浓缩至1 mL,待SPE净化。
2.4.4 净化及富集 固相萃取小柱先后用5.0 mL甲醇和5.0 mL水条件化,上样后加入5.0 mL水淋洗柱体,再用4%氨水甲醇溶液6.0 mL洗脱FQs,收集洗脱液于10 mL玻璃离心管中,50±2℃下氮吹浓缩至约0.3 mL,用流动相定容至1.0mL。
2.4.5 样品测定 将定容样品过0.45 mm的滤膜后,进行HPLC分析,添加标样色谱图见图1(b)。
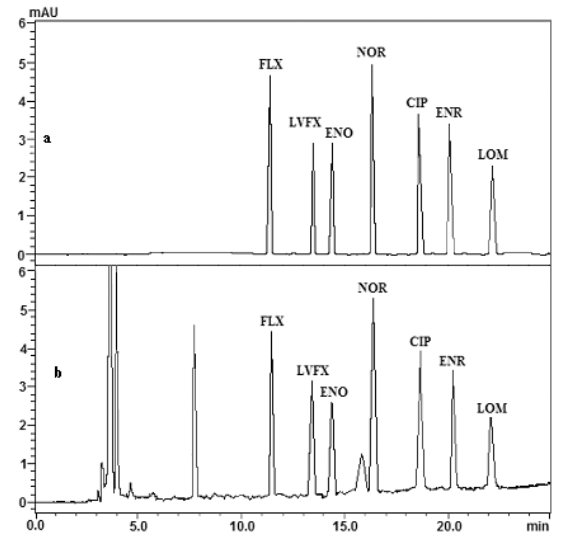
图1 样品色谱图:(a) 氟喹诺酮类标准样品;(b) 添加0.5 mg/kg标样的样品
Fig.1 Chromatograms of sample: (a) Chromatogram of fluoroquinolones; (b) Chromatogram of spiked sample with 0.5 mg/kg of each fluoroquinolone
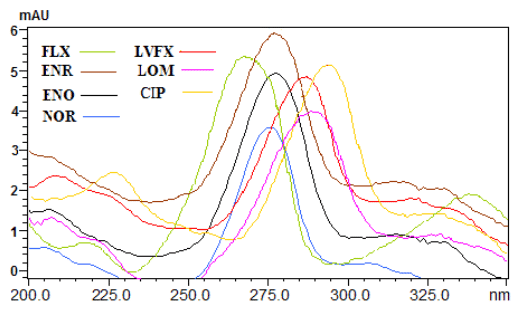
图2 7种氟喹诺酮类药物的紫外光谱扫描图
Fig.2 Ultraviolet absorption spectra of seven fluoroquinolones 3 结果与讨论
3.1 检测波长的选择
取1.0 mg/mL的各单标溶液进液相色谱分析,由二极管阵列检测器可得到各标准样品的紫外吸收光谱图(见图2)。综合考虑各物质的最大吸收波长及吸收强度,我们选定281nm处为检测波长。通过比较样品中各色谱峰的紫外吸收光谱图和保留时间,就可以准确判定其是否含有沙星及其种类。
3.2 流动相的选择
分别用甲醇-磷酸-水、乙腈-磷酸-水两种流动相对7种FQs进行分离发现,各色谱峰形和分离程度没有明显的差别,且乙腈价格高、毒性大,因此选定甲醇系作为流动相。
流速为1.0 mL/min,当甲醇体积分数大于30%时,7种FQs不能完全分离;而小于20%时,保留时间较长且峰形不好;当选用25%时,峰形较好,分离完全且保留时间较理想。
以甲醇和磷酸-磷酸二氢钠-三乙胺缓冲溶液为流动相,通过改变磷酸、磷酸二氢钠及三乙胺的用量调节水系的pH 值,发现磷酸二氢钠浓度在10~20 mmol范围,色谱峰形均较好,因此,选用磷酸二氢钠浓度为10 mmol;固定磷酸的浓度(10 mmol/L),调节三乙胺的用量,考察pH值为2.2、2.6、2.8、3.0发现,pH为2.6、2.8时,7种标样基本分离,而pH为2.8时由于加大了三乙胺的用量,拖尾现象明显改善。所以,选用流动相为:甲醇:10 mmol/L磷酸-10 mmol/L磷酸二氢钠/三乙胺溶液(pH 2.8) = 25:75,流速为1.0 mL/min,此条件下7种FQs能得到很好的分离。文献报道多用乙腈体系作流动相,毒性大,成本高。
3.3 提取溶剂的选择
FQs为两性物质,在稀酸稀碱中溶解性较好[21]。分别用乙腈、乙腈/氨水、乙腈/醋酸作为提取剂发现,乙腈作为提取剂杂质较小,但回收率最低(<60%);乙腈/醋酸回收率最高,因此选定乙腈/醋酸为提取剂。通过改变醋酸的浓度发现,杂质的量随着酸含量的提高而增加,因此选定1%的醋酸/乙腈作为提取剂。
3.4 线性范围及仪器检出限
将混合储备液逐级稀释成标准混合溶液系列:5.0 mg/mL、1.0 mg/mL、0.5 mg/mL、0.1 mg/mL和0.05 mg/mL,在上述色谱条件下分别进行分析,由回归分析得到这几种组分的校正曲线方程(见表1),由表可见这几种物质的峰面积与浓度有良好的线性关系,以0.05 mg/mL测定数据计算3倍信噪比求得仪器检出限为5~10 mg/L。
表1 7种氟喹诺酮类药物校正曲线的回归分析及检出限
Tab.1 Regression analysis of calibration curves and detection limit of seven fluoroquinolones
Compound |
Regression equation |
Correlation coefficient(r) |
Linear range |
Detection limit |
FLX |
Y = (1.40154e-005)X - 0.0405037 |
0.999 9 |
0.05~5 | 7 |
LVFX |
Y = (2.47858e-005)X - 0.0274752 |
0.999 9 |
0.05 ~5 |
10 |
ENO |
Y = (2.15595e-005)X + 0.0171613 |
0.999 9 |
0.05 ~5 |
5 |
NOR |
Y = (1.98659e-005)X - 0.0480307 |
0.999 9 |
0.05 ~5 |
8 |
CIP |
Y = (1.34947e-005)X + 0.0433215 |
0.999 9 |
0.05 ~5 |
7 |
ENR |
Y = (1.40129e-005)X + 0.0637145 |
0.999 9 |
0.05 ~5 |
7 |
LOM |
Y = (1.63983e-005)X + 0.0087081 |
0.999 9 |
0.05 ~5 |
8 |
3.6 回收率、重现性及方法检出限
取空白肉样,向其中准确加入一定量标准混合溶液,按照2.4步骤进行加标回收率实验;加标为0.5
mg/kg和0.2 mg/kg
两个水平,每水平做三个平行样品。回收率、变异系数(RSD%)和定性检出限(LOD)、定量检测限(LOQ)的测定结果见表2。
表2 7种氟喹诺酮类药物在两个水平的加标回收率、重现性及检出限(n=3)
Tab.2 Average recovery, repeatability, LOD and LOQ of
seven fluoroquinolones at two levels of spiking (n=3)
Analyte |
Level |
Recovery(%) |
Mean |
RSD |
LOD |
LOQ |
||
1 |
2 |
3 |
||||||
FLX |
0.5 |
83.1 |
84.0 |
87.0 |
84.7 |
2.4 |
11.0 |
36.7 |
0.2 |
88.6 |
91.9 |
92.8 |
91.1 |
2.4 |
|||
LVFX |
0.5 |
82.6 |
82.7 |
87.6 |
84.3 |
3.3 |
11.7 |
39.0 |
0.2 |
70.6 |
74.3 |
75.6 |
73.5 |
3.6 |
|||
ENO |
0.5 |
68.5 |
71.4 |
73.7 |
71.2 |
3.6 |
9.1 |
30.0 |
0.2 |
63.4 |
64.9 |
69.4 |
65.9 |
4.8 |
|||
NOR |
0.5 |
79.9 |
81.6 |
82.4 |
81.3 |
1.6 |
11.1 |
37 |
0.2 |
77.1 |
78.3 |
83.1 |
79.5 |
4.0 |
|||
CIP |
0.5 |
82.4 |
84.1 |
87.9 |
84.8 |
3.3 |
9.5 |
31.7 |
0.2 |
88.2 |
92.3 |
96.4 |
92.3 |
4.4 |
|||
ENR |
0.5 |
88.1 |
89.6 |
90.8 |
89.5 |
1.5 |
12.7 |
42.3 |
0.2 |
78.3 |
83.9 |
85.6 |
82.6 |
4.6 |
|||
LOM |
0.5 |
82.2 |
85.6 |
85.7 |
84.5 |
2.4 |
13.5 |
45 |
0.2 |
90.1 |
94.3 |
96.1 |
93.5 |
3.3 |
|||
由表2可见,各分析物的加标回收率除依诺沙星(66%~71%)外,其他均在80%~92%之间,变异系数RSD<5%,方法检出限在9~14
mg/kg,接近于荧光检测器的检测水平。
3.7 结论
在选定的条件下,通过对猪肉中7种FQs的回收率、重现性和检出限的分析测定,证明本方法能够有效地同时提取、分离和测定7种FQs的残留量,方法的回收率较高,检出限低,重现性良好,可操作性强,可为相关部门制定FQs类兽药残留的检测方法提供良好的参考。依诺沙星回收率偏低,是本文尚待解决的问题。
REFERENCES
[1] A. Petersen, J.S. Andersen, T. Kaewmak, et al., Environ. Microbiol., 2002, 68: 6036.
[2] Y. Sáenz, M. Zarazaga, M. Lantero, et al., Agents Chemother., 2000,44:
271.
[3] D. Gendrel, M. Chalumeau, F. Moulin, et al., Lancet Infect. Dis., 2003, 3: 537.
[4] Council Regulation (EEC) No. 2377/90 of 26 June 1990 laying down a Community procedure
for the establishment of maximum residue limits of veterinary medicinal products in
foodstuffs of animal origin, Off. J. Eur. Commun. L224, 1990: 1.
[5] 杜黎明, 卫洪清, 张俊燕等. 色谱, 2003, 21: 503.
[6] 刘媛, 谢孟峡, 丁岚等. 分析化学, 2004, 3: 352.
[7] D. Giménez, D. Grasso, L. Sarabia, et al. Talanta, 2004, 64: 442.
[8] C. Ho, D. W. Sin, H. P. Tang, et al. Journal of Chromatography A, 2004, 1061: 123.
[9] M.D. Prat, J. Benito, R. Companó, et al. Journal of Chromatography A, 2004,
1041: 27.
[10] M. Ramos, A. Aranda, E. Garcia, et al. Journal of Chromatography B, 2003, 789: 373.
[11] I. Pecorelli, R. Galarini, R. Bibi, et al. Analytica Chimica Acta, 2003, 483:
81.
[12] M.P. Hermo, D. Barr′on, J. Barbosa. Analytica
Chimica Acta, 2005, 539: 77.
[13] L. A. Shervington, M. Abba, B. Hussain, et al.. Journal of Pharmaceutical and
Biomedical Analysis, 2005, 39: 769.
[14] E. Turiel, G. Bordin,
A. R. Rodr?guez, et al. Journal of Chromatography A, 2003, 1008: 145.
[15] B. Toussaint, M. Chedin, U. Vincent, et al.
Journal of Chromatography A, 2005, 1088: 40.
[16] N. V. Hoof, K. D. Wascha, L. Okerman, et al. Analytica Chimica Acta, 2005, 529: 265.
[17] L. Johnston, L. Mackay, M. Croft. Journal of Chromatography A, 2002, 982: 97.
[18] M. Ferdig, A. Kaleta, T. D.T. Vo, et al. Journal
of Chromatography A, 2004, 1047: 305.
[19] D. Barron, E. Jimenez-Lozano, J. Barbosal. Journal of Chromatography A, 2001,919:
395.
[20] P. G. Gigosos, P. R. Revesado, O. Cadaha, et al. Journal of Chromatography A, 2000,
871: 31.
[21] 国家药典委员会,中国药典(二部),北京:化学工业出版社,2000.