http://www.chemistrymag.org/cji/2006/084026rc.htm |
Apr. 2, 2006 Vol.8 No.4 P.26 Copyright |
Zhang Kainan, Li Yunzheng, Zhang Qingshan,
Guo Bingnan
(School of Chemical Engineering and Environment��Beijing
Institute of Technology, Beijing, 100081)
Keywords Tazobactam; Intermediates; Synthesis
�����̹�����м���ϳ��о��Ľ�չ
������������ѧ�����뻷��ѧԺ ���� 100081��
ժҪ
�����̹��һ������b-������ø���Ƽ������к�ǿ����ø���ԡ����������˽�Щ������������������̹�����м���ĺϳ��о���չ�����Ը��ַ�Ӧ���������˱ȽϺ��ܽᡣ
�ؼ��� �����̹�� �м��壻 �ϳ�
�����̹��tazobactam�����ձ�������ҩ��˾������������ù������b-������ø���Ƽ�����ѧ��3
a-��-7-����-3b-��1H-1,2,3,-����-1-�Ǽ���-4-����-1-����˫��[3.2.0]����-2a-����4��4-�����������������Hall T.W[1]���˴�6-������ù���ᣨ6-aminopenicillianic aicd,6-APA�������Ƶã����Ľṹ�������̹�Ļ���������һ���������������øЧ��������Ŀǰ�ٴ�Ч����ѵ�b-������ø���Ƽ��������ȶ��Ըߣ����Եͣ����Եͣ���ø����ǿ���ص㡣1992�꣬�����̹�ĸ���ҩ�������̹/�������֣�1:8���״��ڷ������У��������ƶ���ϸ����Ⱦ��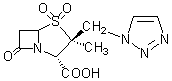
1 �����̹�ĺϳɷ���
�����̹��Ŀǰ�ٴ�Ӧ��Ч����õ�b-������ø���Ƽ�֮һ��Ŀǰ���������ϳɼ��ٴ�Ӧ�õ��о�һֱʮ�ֻ�Ծ�������л��ϳ�ˮƽ�IJ�����ߣ����ĺϳ�·���乤�ղ��ϵõ����ơ������̹�ĺϳɸ��������õ�ԭ�ϲ�ͬ����Ҫ�������ϳ�·�ߣ��ֱ������̹����ù��G���Σ���6-������ù���ᣨ6-APA��Ϊ��ʼԭ�ϡ�
1.1 �����̹Ϊԭ��
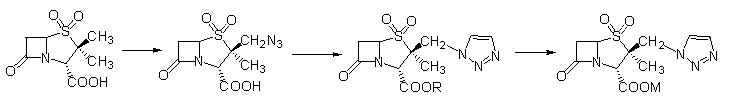
���˲������̹Ϊԭ�ϣ��������������������Ϻ��ѱ����ϳ������̹[2]����ͼ1.1��������·����Ҫ������������ȱ�㣺
��1��ԭ�ϼ۸�ߣ����̹����6-APAΪԭ�ϣ����ص�������������⡢����ȼ�����Ӧ�Ƶõģ����ʽ�Ϊ40%�࣬�г�����ԭ��ҩ��ʽ���ۣ��۸�ϸߡ�
��2������ֱ�ӵ�������Ӧ���ڷ��������ʺܵͣ�����Լ���ϳ�·�ߵĹؼ����ڡ���λ�����ѱ���ȫ�����Ҽ���û������ȡ����������£���������Ӧ�Ƿdz����ѵġ�
��ˣ�����·��ֻ����Ϊ�����о��ḻ�����̹�ĺϳɷ���������������ʵ��������ȥ��
1.2 ����ù��G����Ϊԭ��
Micetich R.G��Maiti S.N����1986�걨��������ù��G����Ϊԭ�Ϻϳ������̹�ĺϳ�·��[3,4]�������ϳ�·�߳�ǰ�IJ���Ӧ�⣬���ಽ������6-APAΪԭ�ϵĺϳ�·����ȫ�Ǻϡ���ǰ�IJ�����ͼ1.2����
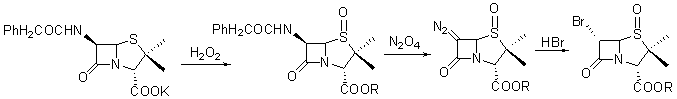
ͼ1.2
��·���ŵ�������ù��G���������6-������ù����۸���˵Ķ࣬ȱ��������ͬ��ʹ����Σ���ױ������ʺ�����ȡ����Ӧ����������Ԫ���칹���������������ǰ���൱��
1.3 ��6-������ù���ᣨ6-APA��Ϊԭ��
��6-������ù���ᣨ6-aminopenicillianic acid, 6-APA��Ϊԭ�Ϻϳ������̹���о���Ϊ��Ծ��Hall T.W.���������6-������ù���ᷨ�Ƶ�[1]��Ŀǰ���ҵ�����������̹�Ĺ���·�ߴ������Ϊ��ʼԭ�ϡ���ҵ����6-APAΪԭ�Ϻϳ������̹��·�ߴ���������[2-5],������ֻ����ǰ������Ӧ���廯��ȣ���Ӧ���Ⱥ�������в�ͬ����·���ܽ���ͼ1.3��
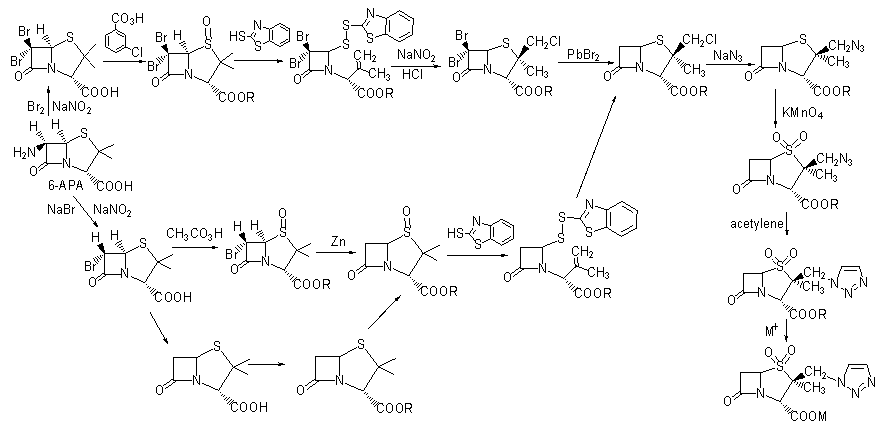
ͼ1.3
2 ���������̹�м����ĺϳ�
2.1 2
2b-�������ù�������������̹�ϳɹ�������ؼ���һ���м���֮һ�����ڴ˲���Ӧ������һֱ�ܵͣ��������ĸĽ��ռ�ܴ�Ŀǰ������������о��ܶ࣬�������������һЩ��ͳ���Լ����µ��о��ɹ���
2.1.1 ��������Ļ��ӳ�
���õĻ��ӳ��Լ���Ҫ�����֣�����Ȳ[1-3��6]�ʹ�����ϩ��[7��8]��
��������ȲΪԭ�ϵĺϳ�·��Ӧ�ý��磬���ŵ�����Ȳ�����ã��������ǶԴ˷�Ӧ�о�Ҳ�ܶ࣬��ҵ������������Ҳ�Ѻܳ��죻��ȱ���Ƿ�Ӧ���ʽϵͣ�ֻ��55%���ң���ʹ�ô�����ϩ���Ե���������л��ӳɿ���ʹ������Ӧ��ߵ�78%����Ӧ�еĴ�����ϩ������������������Ȳ��Ӧ�Ƶã��������ַ�Ӧ�����ڸ�ѹ��Ӧ���ڽ��У�������ȲΪ��ȼ�ױ����壬ʹ��Ӧ����һ����Σ���ԡ�
2.1.2 ֱ�Ӽ�������
1��1-�������-1��2��3-����
���б�������6-APAΪԭ�Ϻϳ������̹�Ĺ����У�ʹ��1-�������-1��2��3-�����ֱ�������������ù���������Ҫ�����м����ӵ�����ͪ���Գƶ������ٵ�������Ŀ�����[9]��
�ݱ������˲���Ӧ�����ʿɴﵽ48%�����ŵ��Ƿ�Ӧ��������ºͣ����������ᵽ�ĵ�������Ļ��ӳ���Ⱥ��߱�����ʹ�ø�Σ���Ե���Ȼ�ձ��Լ���Ȼ��������۸�ԭ��1-�������-1��2��3-����ϳ����ѣ����ʵͣ��Һϳɹ����п��ܷ�����ը�������������������������ȹ��鲻�ܻ��գ����δ����ҵ��������
2�������л������κϳ��������ù������
ʹ�ô����ǹ����������ڷ�Ӧ��ϵ�м���1-��-1��2��3-�������������ܼ��������ֱ����˫���ﷴӦ�����Ƶ�2��-�������ù��������������˷�Ӧ���ŵ��Ƿ�Ӧ���ʺܸߣ��ɴﵽ74%����ȱ�����ڴ˷�Ӧ��Ҫʹ���ж��Ĺ��μ����棬���䷴Ӧʱ��ϳ�����6Сʱ[10]����ͼ2.1.2a��
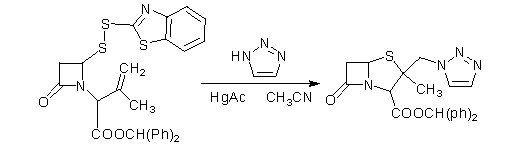
ͼ2.1.2a
������ʹ�úϳ����������̹��·�ߵ��У���һ������������⣬����������2
b-�ȼ���ù�����������������ȡ����Ӧʱ������Ա���ȸ��������ʹ����������ѣ���������Ŀ�����2b--�������ù��������������ʵͣ�����Ӱ���������̹�ϳɵ������ʣ�ʹ��ɱ��ܸߡ��ݱ���[11]��ʹ����������������ӵ�����ͪ�IJ��Գ�˫�������Ӧ��ֱ������2b-�������ù����������������Ҵ˷�Ӧû����Ա���칹���������ɣ�ͼ2.1.2b�����˷�Ӧ�м���һ�����ĵ⣬ʹ�����������е����ܿ��γɳ������Ӷ���ǿ����������ԡ��ݱ������˲���Ӧ���ʿɴﵽ75%����ʵ���˹ػ���������ȡ��һ����ɣ����տɴ����������̹�����ʣ������������˱���[11]�����ô˷���������ʹ�����̹����������ߵ�54%����������Ϊֹ�������������̹�ϳɵ�������ʡ���ͼ2.1.2c��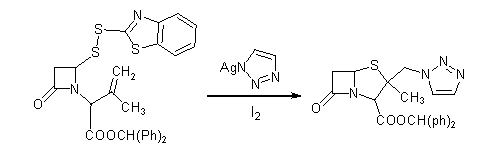
ͼ2.1.2b
ͼ2.1.2c
2.2
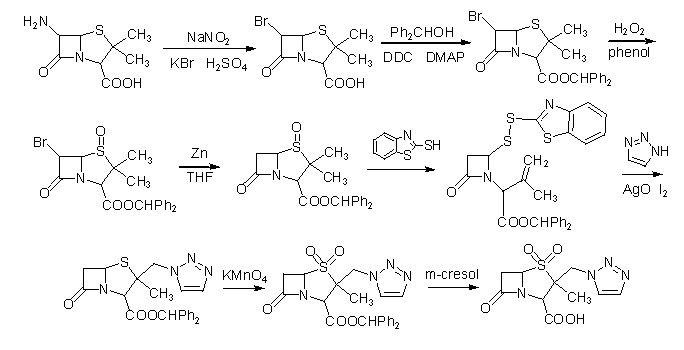
���������ù����������������ĺϳ�
���廯�������������Ǻ���������Ӧ������ġ��ɲ��õ���������˫��ˮ���������ᡢ���ȹ��������ᡢ�ߵ����ơ��������̡�������[12-19]�����������ù���������������������Ҳ�Ƕ��ֶ�����������Ҫ���ܹ�ҵ��������������Ҫʹ�õķ����Լ����ڽ��Ľ�������DZ�����õ��·�����������Ӧ��ͼ2.2��
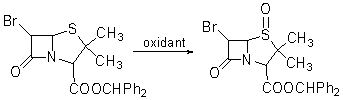
ͼ2.2
2.2.1 ��ͳ����
1�� ���ȹ���������(m-CPBA)��
�������̹�Ĺ�ҵ���������̵��У����ȹ��������������������õ������������������ڵı����������ù������������ɱ����ȹ��������ᣨm-CPBA���������������ù������������������˷�Ӧ��m-CPBA
Ϊ�������������Ϊ�ܼ��������¼��ɽ��У���Ӧ70���ӣ����ʿɴ�90%��
2�� �������ᷨ
����������ȹ������������Ҫ���˵öࡣʹ�ù�������Ϊ������ʱ����Ũ�ȶԷ�Ӧ��Ӱ��ܴ�Ronald����[4]����40%�Ĺ�������Ϊ����������6��-�����ù�������������������90%���ϣ��������������Ĺ���������Ʊ�������ȫ�ֲ����ã������ȶ��Բ������ù�����������������������������ù�������������,������������Ӧ�����õ����������Ĺ������ᡣ���ε�����˱���[19]����6%?%�Ĺ�����������������Ҳ�ɴﵽ75.2%��
2.2.2 ���һЩ�µ���������
1�� ˫��ˮ���������������HFIP��
�˷�����Ravikumar, K. S.����[12]���ܸ�Ч�ؽ�һ����������Ϊ���������˷�Ӧ�õ������̹�ĺϳɵ��У������������Ϊ�ܼ�������������Ϊ30%��˫��ˮΪ�����������½��������������������������˷�Ӧ�����������������ɡ���ȱ��������������۸��Դ�����е�ͣ��ӷ��Դ������ѡ�
2�� ˫��ˮ������/�������ӷ�
�����ױ�����ʹ��˫��ˮΪ�����������ӻ���������Ϊ�ܼ������Ը�Ч��ѡ���ԵĽ���������������[20-21]��
�˷������������̹���������б��dz��õ����ã�6
3�� ����������
�˷���Martin S E��Rossi L I [22]��2001�����ȱ���������Fe(NO3)3-FeBr3 Ϊ����������Ϊ�ܼ�������Ϊ��������������������Ϊ�������˷��������뵽�����̹�ĺϳ���[23]���ݱ�����ͬ�����������ʹ������ڵ�����£�ʹ�ü��Է����ӵ�DMFΪ�ܼ������Ը�Ч�Ľ�2��-����ù�����������������2��-����ù�������������������������������90%�������������Ƚ����룬��Ӧʱ��̩p�ɱ��ͩp���ʸߣ�����û����Ⱦ�IJ�����������ɫ��ѧ�ľ��������ԭ�Ӿ����ԡ���������DMF�нϸߵ�����ѹ���е�Ҳ�ϸߣ���ʹ�������㣬��Ϊ������������ȱ�����ڡ�
4�� IBX��
�ߵ��Լ����л��ϳɻ�ѧ�е�Ӧ�ã��Ѿ�����㷺�Ĺ�ע���ߵ�����Ӧ�������������������ķ�Ӧ�����б�����Ȼ�������ڶ�ߵ��Լ��У�1-�ǻ�-1��2-������-3��1H��-ͪ-1-�����IBX�������º͵ġ�ѡ���Եķ�Ӧ�����Ѿ��ڶ����л��ϳɷ�Ӧ�еõ���Ӧ�ã�ͬʱ���������еͶ��������ڶ����ܼ����������ŵ㡣��IBX�����������ɽ��������ù������������������������[24]����Ӧ�������½��У��ڷ�Ӧ��ϵ�м������һ��廯�����������Ӧ�ڰ�Сʱ����ɣ�����ֻѡ���Ե�������������û�����Ĵ��ڣ����ʿɴﵽ89%��
2.3 ���������ù����3λ�Ȼ��ı�������
Ϊ��ֹ���ȷ�Ӧ�ķ����Լ��ں�����Ӧ���Ȼ��п��ܷ�����������Ӧ���Ȼ������DZ���ġ������Լ���ѡ���ϣ�����Ҫ���Dz�Ʒ�к���һ����ˮ�����е���-����������Ϊ�˷�ֹ�����ѱ���ʱ������ֻ��ѡ��������л����Լ������л����Լ������������ѱ��������º͵Ļ������������ᱣ������α����������ͼ��ԣ��ڻ�ѧ��Ӧ�н�Ϊ�ȶ���������һЩ��Ϊ���õı����Լ���
2.3.1 ʹ�ö������л�����
�������л��DZ����Ȼ�����Ҫ�Լ����������̹�ĺϳ��У���ʹ�ö��������Ȼ��б���3λ�Ȼ����˷�Ӧ�������¼��ɽ��У�ʹ��N��N-������������DMAC��Ϊ�ܼ������ʿɴﵽ70%[25]��
2.3.2 ʹ�ö����걣��
���ȶ��Ķ����������������Ȼ������ķ����Ƕ��������ù����3λ�Ȼ����б�������÷���[26]���˷�����Ϊ���죬��������ڹ���������ڵ�������Ǻϳɹ����ж���1λ���ѵ������Ͷ�3λ�Ȼ��ı���ͬʱ���У��������������������ʿɴ�90%��ʵ��֤������Ӧ�м����KI �ڴ˷�Ӧ�б�����Ϊ�ߵ�����������������á�ʹ�ö������ȱ�����ڣ�һ���������۸ɱ��ϸߣ���һ������������Ӧ�����������ᴿ�����˲���[27]��
2.3.3 �Զ����״�Ϊ�����Լ�
�����״�����ڶ�������Լ۸���Ե�������Ӧʱ��̣������ºͣ���Ӧ���ʸߡ��������̹�ĺϳɵ�����һ��ʮ����Ҫ�ı����Լ�[6]���ö�������̼���ǰ���DCC��dicyohexlcarbodiimide����Ϊ�������ϼ�������DCC�����Ʊ�2b-����ù�����������ʱ����Ӧ�ڶ�ʱ������ɡ�����ʹ�ö��װ�����ण�DMAP����DMAP�ڷ�Ӧ��ϵ�����л�������ã����ļ���ʹ��Ӧϵͳ�������ӣ���Ӧ�������½��У�˲����ɣ����ʽӽ�100%[11]��DCC����һ������������������ر�������2b-����ù��������λ��ϴ��������л��ŵ���Ͷ����״������������������ǻ��γ��ȶ������ӵĴ�֮���������
ͼ2.3�ǶԸ����Ȼ������������ܽ
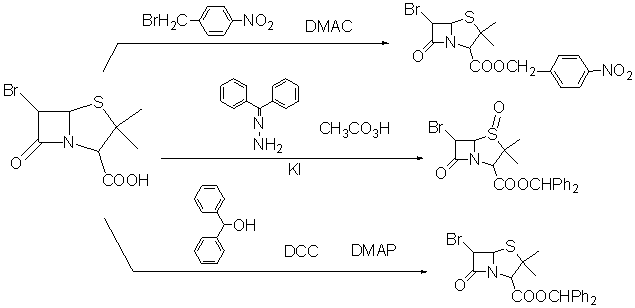
ͼ2.3
3 ����
���������̹������ٴ����ܣ����������ϳɽ����˹㷺�о�����Ŀǰ��Ϊ�ձ�����������У������̹���������ʽ�Ϊ12%�����Խ�һ���Ľ������̹�ĺϳɹ��գ����������Ѿ���Ϊ����֮����Ȼ������ҲӦ�����������̹����øЧ����Ȼ���ڿ���ά������̹�������ͷ�߾���ø������Ч��Ҳ�������ԡ���ˣ����о��Ľ������̹�ϳ�·�ߣ�������ɱ���ͬʱ�����������µĸ�Ч������
REFERENCES
[1] Hall T W, Maiti S N, Micetich R G, et al. Royal Society of Chemistry, 1985, 22:
242-245.
[2] Zhang Jianmin��Zhong
Chaoping. Chinese Journal of Pharmaceuticals, 1995��26��9����428-429.
[3] Micetich R G, Maiti S N, Spevak P, et al. J Med Chem, 1987, 30(8): 1469-1474.
[4] Micetich R G, Maiti S N, Spevak P, et al. Synthesis, 1986, 4: 292-296.
[5] Tanaka H, Tanaka M, Nakai A, et al. Bull Chem Soc Jpn. 1989, 62(2): 627-629.
[6] Hu Yuehua��Hu Yimin.
Chinese Journal of Synthetic Chemistry��2003��1(11):
243-245.
[7] ɽ��ï. �ձ�����������B2��. ƽ45034,
1992-01-30.
[8] Micetich R G. EP 138,523. 1985-04-24.
[9] Toriis S, Tanak H, Tanka M, et al. EP, 331395, 1989-09-06.
[10] Torll S, Tanaka M, et al. US4898939, 1990-02-06.
[11] Wei Liang Xu, Yun Zheng Li, Qing Shan Zhang. Synthesis, 2005, 3: 442-446.
[12] Ravikumar K S, Barbier F, Bégué J-P, et al. J Fluorine Chem, 1999,
95: 123-125.
[13] Vajender S V, Saini R K, Maeshram H M. Tetrahedron Letters, 1997, 38: 6525-5628.
[14] Martín S, Rossi L I. Tetrahedron Letters, 2001, 42: 7147-7151.
[15] Crich D, Neelamkavil S. Tetrahedron, 2002, 58: 3865-3870.
[16] Asensio G, Mello R, González-Nú?ez M E. Tetrahedron Letters, 1996, 37:
2299-2302.
[17] Sato K, Hyodo M, Aoki M, et al. Tetrahedron, 2001, 57: 2469-2476.
[18] Lindén A A, Krüger L, B?ckvall J-E. J Org Chem, 2003, 65: 5890-5896.
[19] Song Danqing, Wang Lulu, Zhang Zhiping, et al. Chinese Journal of Pharmaceuticals,
1999, 30(1): 35-39.
[20] Wei Liang Xu, Yun Zheng Li, Qing Shan Zhang, He Sun Zhu. Synthesis, 2004, 2: 227-233.
[21] Wei Liang Xu, Yun Zheng Li, Qing Shan Zhang, He Sun Zhu. Synthetic Communications,
2004, 34(2): 231-237.
[22] Martín S, Rossi L I. Tetrahedron Letters, 2001, 42: 7147-7151.
[23] Wei Liang Xu, Yun Zheng Li, Qing Shan Zhang, He Sun Zhu. The Journal of Beijing
institute of Technology, 2004, 13, Suppl: 88-90.
[24] Xu Weiliang, Li Yunzheng, Zhang Qingshan, Zhu Hesun. Fine Chemicals, 2004, 4:
304-306.
[25] Willian J G, Leonard B. Crast Jr, Robert G G, et al. J Med Chem, 1981, 24: 1531-1534.
[26] Osborne N F, et al. J Chem Soc Chem Commun, 1989, (6): 371-373.
[27] Cao Lin, Chen Ligong, Li Yang, et al. Chemical Industry and Engineering, 2002, 19(3):
219-224.