http://www.chemistrymag.org/cji/2006/084028pe.htm |
Apr. 2, 2006 Vol.8 No.4 P.28 Copyright |
(College of Chemistry and Environmental Science, Hebei University, Key Laboratory of Analytical Science and Technology of Hebei Province, Baoding 071002, China) Received Jan. 18, 2006. Abstract Derivative molecular spectrophotometry is based on the determination of variation rate of signal intensity with wavelength (dI/dl). A new derivative technique based on the determination of variation rate of signal intensity with time (dA/dt) has been developed for vapor generation atomic absorption spectrometry. The models of conventional signal and the equations of derivative signal are described for cold vapor atomic absorption spectrometry (CVAAS) and hydride generation atomic absorption spectrometry (HGAAS). The principle and performance of the vapor generation atomic absorption spectrometry using derivative signal processing are evaluated. The intensity of derivative signal is directly related to the concentration of analyte with a good linearity. In comparison with conventional vapor generation atomic absorption spectrometry, the method has higher sensitivity and lower detection limit. The applications of the derivative vapor generation atomic absorption spectrometry in trace analysis are reviewed.
Keywords Vapor generation��Atomic absorption spectrometry��Signal models��Derivative measurement principle��Trace analysis 1 INTRODUCTION
Derivative molecular spectrophotometry is based on the determination of variation rate of signal intensity with wavelength (dI/dl). Bosch et al. reported all the methods that were currently available to researchers for utilizing derivative ultraviolet/visible absorption spectrophotometry and their analytical applications until 1993.[1] The recent publications were reviewed on theoretical aspect of derivative spectrometry and its use in chemical analysis, pharmaceutical analysis, food analysis, clinical analysis and other field of applications published since 1994.[2] The spectrum obtained with derivative spectrophotometry offers a convenient solution to a number of well-known analytical problems��such as the resolution of multi-component systems��removal of sample turbidity��matrix background and enhancement of spectral details.
Atomic absorption spectrometry is an accepted and widely used method for the determinations of micro elements in a great variety of samples. But its sensitivity could not meet the demands of trace and ultra-trace analysis for some sample. Analysts have studied on how to enhance the sensitivity of flame atomic absorption spectrometry by various ways, and most of the studies focus on the improvement and application of preconcentration technique by using chemical and physical methods. All of the reported methods are based on the measurement of signal intensity and there is no any breakthrough in the measurement technique. A new derivative technique based on determination of variation rate of signal intensity with time (dA/dt) has been developed by Sun's group for flame atomic absorption spectrometry (FAAS).[3] This derivative technique is different from the derivative molecular spectrophotometry based on the determination of variation rate of signal intensity with wavelength (dA/dl). The sensitivity of Cu, Mn, Fe, Zn, Cd and Pb for the derivative flame atomic absorption spectrometry (D-FAAS) can be remarkably improved at 2mVmin-1with 50 times higher as compared with FAAS.[4] The applications of the derivative flame atomic absorption spectrometry in trace analysis have been reviewed.[5]
Application of hydride generation for atomic spectrometry is the most efficient method for the determination of hydride-forming elements. Many methods coupled with hydride generation, such as HGAAS, HGAES, HGICP and HG-MS, are effective, but for an accurate determination of trace element in water, pre-concentration is generally required. The proposed derivative technique has been applied to cold vapor atomic absorption spectrometry (CVAAS) and hydride generation atomic absorption spectrometry (HGAAS) for the determinations of trace elements in biological and environmental sample.[6-8] The purpose of this paper is to review the methodology and application of the derivative vapor generation atomic absorption spectrometry.
2 SIGNAL MODEL
2.1 Signal model for HGAAS
The signals of conventional and derivative vapor generation atomic absorption spectrometry are shown in Fig.1. [9]
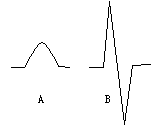
Fig.1 The conventional (A) and derivative signals (B) for vapor generation atomic absorption spectrometry
A conventional signal for
CVAAS and HGAAS is similar to a pulse signal to be used for analysis. A derivative signal
for CVAAS and HGAAS consists of an up-peak and a down-peak connected with the end and the
head.
The model of conventional signal for HGAAS was given as follows:[9-11]
for up-side: Au= A0·[ 1-exp(-ta /b)]
for down-side: Ad = A0· exp(-tc/d)
Where a, b, c and d for any element are constant in a
wide range of concentration. It was given in Table 1.
Table 1 The values of a, b, c and d for studied elements
Element |
a |
b |
c |
d |
Bi |
1.80 |
8.08 |
1.01 |
26.74 |
As |
2.0 |
2.16 |
1.45 |
17.00 |
Se |
1.10 |
6.32 |
1.18 |
20.66 |
Sb |
1.62 |
7.66 |
1.02 |
24.68 |
Te |
1.98 |
6.44 |
1.02 |
24.68 |
Sn |
1.01 |
7.14 |
1.58 |
24.56 |
Pb |
1.62 |
6.99 |
1.01 |
22.79 |
The variation of the derivative signal
intensity with time for derivative HGAAS was obtained by derivativing the model to time,
respectively.
for up-side��
for down-side��dAd/dt=c·d-1·A0·tc-1·exp(-tc /d)
2.2 Signal model for CVAAS
Generation of Hg vapor is based on reaction of Hg2+ with NaBH4.The change rate of mercury quantity at the inlet of absorption cell can be expressed as the difference of the rate of introduction n1(t) and that of dissipation n2(t), it was expressed as dnA/dt = n1(t) - n2 (t).The redox reaction of generation of mercury vapor could be considered as a first-order reaction if the acidity of medium was a constant and the quantity of potassium tetrahydroborate was excessive. Then the introducing rate of mercury vapor at any time can be expressed by n1(t) = k2·k1· n0·exp(k1t) , where k1 is the reaction rate constant, n0 is the initial quantity of mercury ion(II), and k2 is a proportion factor which is related to volume of test solution and length of conduit. The dissipation rate of mercury vapor from the cell at any time can be given by n2(t) = k3·(FD/��r2 )· nA, where k3 is a proportion factor, F is the flow rate of carrier gas , r is the radius of absorption cell , and D is the diffusion coefficient of mercury atom, nA is the quantity of mercury vapor in the place of inlet at any time. Then dnA /dt was given by [6]
dnA/dt = k2 k1 n0 exp(k1t)��k3 (FD/��r2 ) nA
The quantity of mercury vapor at different points in the cell can be given by n = nA (l-x / L), where L is the length of the absorption cell and x is diffusion distance of mercury vapor. The total quantity of mercury vapor has been obtained, then the model of absorbance signal was given by
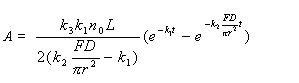
The equation for the derivative CVAAS has been obtained by derivativing the model to time.
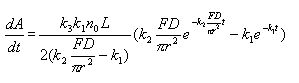 3 PRINCIPLE AND
PERFORMANCE
3 PRINCIPLE AND
PERFORMANCE 3.1 Derivative analysis principle
The laboratory-made derivative measurement system consists of a magnification and a differential unit. [8] The circuit diagram of derivative measurement equipment is shown in Fig.2.
An output signal has a rigorous derivative relation with the input ones. The output signal of the system will keep in base-line when the variation of the input signal is zero, and when the variation of the input signal is not zero, there is a corresponding polar output which is in direct relation to the variation of the input signal. The derivative measurement equipment was connected between an atomic absorption spectrometer and a double-pen recorder, as shown in Fig.3. The derivative and conventional signals were recorded simultaneously with the double-pen recorder.
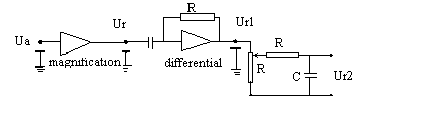
Fig.2 The circuit diagram of derivative measurement equipment
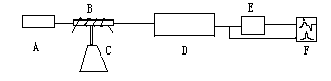
Fig.3 Derivative atomic absorption spectrometric system
A: Lamp-house, B: Atomizer, C: Hydride generation equipment, D: Spectrophotometer, E: derivative measurement equipment, F: Recorder
Based on
differential principle, the intensity of output signal of the derivative measurement
system was expressed as D = -m·B·RC·dA/dt, where m is magnification multiple of
magnification unit, RC is time constant of differential unit, B is
sensitivity of differential unit. The height of up-peak and down-peak of derivative signal
are expressed as Du and Dd, respectively.
The total height of derivative signal is expressed as D, where D= Du + Dd.
The equations for up-peak and down-peak of derivative signal were obtained.[8]
The total height of the up-peak and down-peak is expressed
as: [6-8]
for D-HGAAS: 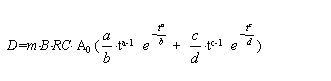
for D-CVAAS: 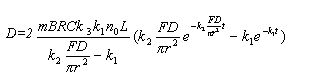
For the derivative system, both RC and m
are a constant. As A0=k·c for HGAAS, and
CVAAS used peak time as measurement time, the intensity of derivative signal can be
expressed as D =K·B·c. When derivative measurement sensitivity (B)
was selected, the intensity of derivative signal is directly related to the concentration
of analyte. It provided theoretical principle for derivative atomic absorption
spectrometry.
3.2 Analytical performance
The curve drawn based on the mathematical model is in good agreement with the signal
measured experimentally. The test result showed that the proposed derivative atomic
absorption spectrometry has a good linearity for the determination of trace and
ultra-trace element, as shown in Table 2. The sensitivities for the derivative atomic
absorption spectrometry can be remarkably improved at 2mVmin-1with 16-72 times
higher as compared with vapor generation atomic absorption spectrometry, as shown in Table
3, along with the relative standard deviation range at 20, 10, 5 and 2 mV min-1.
The detection limits of derivative vapour generation atomic absorption spectrometry and
derivative spectrophotometry(DS) for the determinations of some elements are given in
Table 4. In comparison with derivative spectrophotometry, the derivative vapour generation
atomic absorption spectrometry has higher sensitivity and lower detection limit, but it
was used only to the determination of single metal element.
Table 2 Regression equations and correlation coefficients [7]
Element |
Sensitivity range (mV min-1) |
Regression equation* |
Linear
range |
Correlation
|
As |
20 |
A=0.1700 C - 0.00002 |
0-100 |
0.9998 |
10 |
A=0.3450 C + 0.00002 |
0-100 |
0.9994 |
|
5 |
A=0.9293 C + 0.00004 |
0-100 |
0.9991 |
|
2 |
A=1.9860 C + 0.00006 |
0-100 |
0.9959 |
|
Sb |
10 |
A=0.1924 C + 0.00005 |
0-100 |
0.9996 |
2 |
A=1.1160 C - 0.00005 |
0-80 |
0.9986 |
|
Bi |
10 |
A=0.3414 C - 0.0004 |
0-100 |
0.9997 |
2 |
A=1.8180 C + 0.0001 |
0-80 |
0.9998 |
|
Sn |
10 |
A=0.1441 C -0.0005 |
0-100 |
0.9992 |
2 |
A=0.6693 C + 0.0002 |
0-80 |
0.9989 |
|
Pb |
10 |
A=0.0487 C +0.00002 |
0-150 |
0.9988 |
2 |
A=0.2405 C +0.00001 |
0-150 |
0.9985 |
|
Te |
10 |
A =0.0198 C +0.0002 |
0-200 |
0.9981 |
Hg |
2 |
A=0.024 m -0.0002 |
1.8-35.0 ng |
0.9968 |
* A: derivative absorbance, C: mg L-1, m: mass, ng
Table 3 Improved fold of sensitivity and detection limit and relative standard deviation
Element |
Sensitivity range (mV min-1) |
Increased
sensitivity |
Improved detection limit (Fold) |
RSD(%) |
Pb |
2 |
26 |
14 |
3.8 ~ 4.8 |
Se |
2 |
34 |
16 |
2.4 ~ 4.2 |
Te |
2 |
52 |
37 |
4.2 ~ 5.3 |
As |
2 |
36 |
13 |
1.7 ~ 4.8 |
Bi |
2 |
47 |
8.6 |
2.6 ~ 4.8 |
Sn |
5 |
16 |
12 |
2.2 ~ 5.2 |
Sb |
2 |
28 |
8.9 |
2.1 ~ 4.2 |
Hg |
2 |
72 |
24.5 |
2.8 |
Table 4 Comparison of detection limits between derivative vapour generation atomic absorption spectrometry and derivative spectrophotometry(DS)
Method |
Order |
Analyte |
Sample |
Linear range |
Detection limit(mg L-1) |
Ref. |
DS |
second |
Hg |
Waste water |
18-104 |
18 |
[12] |
DS |
First |
Se |
Tea |
0-1000 |
0.13 |
[13] |
DS |
First |
Se |
Water, blood, Hair |
0-250 (ng) |
1.5 (ng) |
[14] |
DS |
First |
Cd, Hg |
Sythetical sample |
420-9200 | 34, 34 | [15] |
DS |
First |
Pb, Cd, Hg |
Sythetical sample |
0-2, 0-40, 9-200 |
4, 2, 10 |
[16] |
DCVAAS |
First |
Hg |
Cosmetic sample | 1.8-35.0 (ng) | 155 (pg) | [17] |
DHGAAS |
First |
Pb |
waters |
0-150 |
0.096 |
[10] |
DHGAAS |
First |
Te |
Urine | 0-200 | 0.26 | [18] |
DHGAAS |
First |
Se |
Urine |
0-150 |
0.074 |
[19] |
Tellurium is a poisonous element in living biological systems, it can be accumulated in kidney, heart, liver and spleen, and induce the degeneracy of liver and kidney in excess of 0.002 g kg-1. [20] The content of tellurium in kidney, liver and muscle is 0.07, 0.014 and 0.017 mg kg-1, respectively.[21] In view of the extremely low concentration in biological samples, it is necessary to develop sensitive, precise and accurate analytical methods for tellurium determination in biological specimens. Kobayashi and Imaizumi determined tellurium in urine by graphite furnace atomic absorption spectrometry after solvent extraction and hydride generation atomic absorption spectrometry.[22,23] Siddik and Newsman published an electrothermal atomic absorption spectrometry (EAAS) method using platinum as a modifier for tellurium determination in urine, plasma and tissues.[24] Because of the volatile nature of tellurium, a suitable chemical modifier should be selected when using EAAS. A new method has been developed for the determination of tellurium in urine by hydride generation atomic absorption spectrometry with derivative signal processing (D-HGAAS).[18] Urine samples of 100 ml each were digested in the beakers on an electric hot-plate with the consecutive addition of 20 ml of concentrated HNO3 and 10 ml of HClO4. The samples were digested for about 1 h until the appearance of white crystals, then the excess of HNO3 was removed by heating. The beaker was removed and cooled down. Residues were dissolved with 10 ml of 1% HNO3 and diluted to 100 ml with sub-boiling distilled water for analysis. The proposed method had been applied to the determination of tellurium in urine samples from a small population of normal individuals with a recovery range of 89-98%. The characteristic concentration (gives a derivative absorbance of 0.0044) and the detection limit (3s) for tellurium were 0.042 and 0.26mg L-1, respectively, 52 and 26 times better than those of conventional HGAAS. The precision of this method, expressed as RSD, was in the range of 3.5-5.1% for samples of 0.64-1.82mg L-1.
The recommended daily dietary allowances of selenium for woman and men are 55 mg and 70 mg per body, respectively.[25] But at higher concentrations selenium is toxic. Because of the biological importance of selenium in living biological systems, the hydride generation atomic absorption spectrometry was used for the determination of selenium in serum with detection limit of 0.40mg/L and RSD of 2126%-3176%,[26] and in urine with detection limits of five selenium compounds were between 3 and 8 mg L-1, and the relative standard deviation of <7% at 100 mg L-1Se.[27] Derivative hydride generation atomic absorption spectrometry has been developed for the determination of selenium in urine,[19] using sample treatment procedure described in literature.[18] The characteristic concentration and the detection limit (3s) were 0.014 and 0.0074mg L-1, respectively. The sensitivities were improved by 3.6, 6.6, 13.1, and 51.0 fold, and the detection limits were improved 2.2, 3.0, 5.3, 21 times beyond those of HGAAS at 20, 10, 5 and 2 mVmin-1 sensitivity setting, respectively. The derivative HGAAS has a good precision with RSD of 2.2-2.4%.
Low level mercury determination and the evaluation of mercury pollution have received great attention owing to its high toxicity and pollution. Because of its simplicity, high sensitivity and relative freedom from interferences, cold vapour atomic absorption spectrometry(CVAAS) has generally been used for the determination of mercury in various samples. Some methods for the determination of mercury have been developed using a combination of cold vapour generation, and trapping in a porous gold-plated graphite mini-tube, [28] a gold-coated[29,30] or a platinum-lined graphite tube,[31] followed by atomic absorption spectrometric detection. Flow injection CVAAS was used to determine Hg in food with detection limit of 1.2mg/ kg and RSD of 4.7-8.4 %.[32]
A cold vapor atomic absorption spectrometry with derivative signal processing has been developed for the determination of total mercury.[17, 33] Standard reference materials and cosmetic samples were deposited in PTFE pressure digestion vessels for the procedure described below. 1 g of cosmetic sample or 2 g of standard reference material sample was weighed accurately in the PTFE container and mixed with 3 ml of concentrated nitric acid and 0.5 ml of concentrated perchloric acid. The vessels were then covered with PTFE covers and put aside overnight. The vessels were placed in stainless steel bombs, which were sealed tightly with a screw closure to avoid gas leakage, and placed in an oven. The oven temperature was raised to 170ºC over 0.5 h and kept at this temperature for 7 h. The bombs were taken out of the oven and cooled to room temperature. The PTFE vessels were taken out of the stainless steel bombs and the covers were removed. The vessels were heated on a hot plate at 100-110ºC and the solutions were evaporated to about half of their initial volume. After cooling to room temperature, the solutions were transferred to a 25 ml volumetric flask and diluted to the mark with 0.2 mol L-1 hydrochloric acid. The conventional and derivative signals of mercury atoms were recorded with the doublepen recorder. The sensitivity and detection limits of derivative CVAAS were 192 and 155 pg, respectively, 72 and 24.5 times better than those of conventional CVAAS. Total mercury in standard reference materials, mussel (GBW08571) and peach leaf (GBW08501) was determined by the proposed method, and the obtained values were consistent with the reference values. The proposed method had been applied to determine total mercury in skin lotion, astringent lotion, massage cream, cream E-C, and washing milk samples with a recovery range of 92-102%and RSD of 2.8%.
The determination of trace elements has received increasing attention in environmental pollution studies. In particular, there is an increasing need for a simple, sensitive and accurate method for determining sub-parts-per-billion levels of elements in environmental waters. Elements forming covalent hydrides such as antimony, arsenic, bismuth,selenium, tellurium,tin, lead, etc., are often determined by after hydride generation atomic absorption spectrometry. But for an accurate determination of trace element in waters, pre-concentration is generally required. A hydride generation atomic absorption spectrometry under sub-atmospheric pressure was described for determination of Sb, As, Bi , Se ,Te and Sn, with the sensitivities of 290,80, 90, 73, 140 and 110pg, and detection limits of 610, 100, 50, 60, 26 and 200pg, respectively.[34] A continuous flow injection hydride generation atomic absorption spectrometry was described for determination of major antimony species in seawater using pre-concentration techniques. After continuous flow injection hydride generation and collection onto a graphite tube coated with iridium, antimony was determined by graphite furnace atomic absorption spectrometry. The low detection limits were about 5 ng L-1 for Sb(III) and 10 ng L-1 for Sb(V) for 2.5 ml seawater samples.[35]
The derivative hydride generation atomic absorption spectrometry with signal processing had been developed for the direct determination of trace and ultra-trace level of As, Sb, Bi, and Sn in water samples.[11, 36, 37] The effects of atomization temperature, argon flow rate, acidity and concentration of KBH4 and KI were investigated and analytical conditions were optimized. The sensitivities for arsenic and antimony were increased by 36.4 and 27.6 times better than those of HGAAS. For a 2 mV min-1, sensitivity range setting, the characteristic concentration was 0.003mg L-1 for arsenic and 0.004 mg L�C1 for antimony, and the detection limits (3s) were 0.015 mg L�C1 for arsenic and 0.020 mg L�C1 for antimony. The proposed method was applied to the determination of arsenic and antimony in tap water, lake water and mineral water samples with recoveries of 93�C110% and RSD of 3.0�C5.0%.[11] The proposed method has been applied to the determination of bismuth and tin in tap water, lake water, well water and mineral water with recovery of 100% and 95.4% for bismuth and tin, respectively, and RSD of 3.0-5.0%[36]. The calibration curves were linear in the range of 0-100ng·ml-1 for Bi and 0-80ng·ml-1 for Sn with correlation coefficients of 0.9989 to 0.9999. For a 2 mV min-1 sensitivity range setting, the characteristic concentration was 0.003 mg·L-1 for bismuth and 0.004mg·L-1 for tin, and the detection limit (3s) was 0.012mg·L-1 for bismuth and 0.010 mg·L-1 for tin, respectively. The sensitivities were increased by 42- and 31-fold and detection limits were improved by 10-fold for Bi and Sn when using sensitivity range of 2 mV min-1, respectively, as compared to HGAAS. Used boracic acid as the reaction medium, the sensitivity for tin was increased 43 times better than those of HGAAS.[37] The relative standard deviation of 7 replicate determinations of tin in water samples at 0.08-0.72mg·L-1 was 3.9% to 4.7%. A new DHGAAS method was applied to the determination of traces of lead in waters.[10] The detection limit and sensitivity of the proposed method were 26 times and 8.8 times better, respectively, than those of conventional hydride-generation atomic absorption spectrometry. The characteristic concentration (for a derivative absorbance of 0.0044) and detection limit (3s) for lead were 0.017 and 0.096 ng ml�C1, respectively, for a 2 mV min�C1 sensitivity range setting. The recovery range was 92.5-103%.
Lead poisoning has received considerable attention in recent years because of its high toxicity. The prolonged intake of even low concentration of lead can cause serious toxic effects. It is important to develop sensitive method for determination of trace lead in food and biological sample. GFAAS is the most efficient method for the determination of lead traces with the disadvantage of higher cost and greater susceptibility to chemical interferences. HGAAS has been proved an extremely useful method. A combination method of microwave assisted digestion-plumbane generation atomic absorption spectrometry for the determination of lead in foodstuff s was described. Sensitivity of the method was 0. 065mg L-1, detection limit was 0.070 mg L-1, and RSD was better than 3 % with average recovery of 97 %.[38] A method of hydride generation - atomic absorption spectrometry was described for the determination of trace amounts of lead in salt and salt products with detection limit of 0.025 mg kg-1 Pb.[39] However, the conditions for lead hydride generation are critical and the sensitivity is bad compared with that for other hydride-forming elements.
A new method is developed for the determination of trace level of lead in seasoning by derivative hydride generation atomic absorption spectrometry.[40] At temperature of 900ºC, carrier gas flow rate of 600 ml min-1, medium acidy of 1.0% H2SO4 were selected with the maximum derivative absorbance. The effect of different oxidants on hydride generation was examined. The derivative absorbance was zero without oxidant and increased after addition of a kind of oxidant. The derivative absorbance value changed unmistakably with the concentration of KCr2O7 and H2O2 without reaching a plateau. The derivative absorbance increased substantially after addition of K3Fe(CN)6 and became constant when the concentration of K3Fe(CN)6 was higher than 0.5%. 1% K3Fe(CN)6 and a 5 ml of solution volume was chosen for hydride generation. The derivative absorbance measured at 20, 10 and 5 mV min-1 with a 5 mg L-1 of lead solution were 0.1313, 0.2690 and 0.4706, respectively, and 0.4913 with a 2 mg L-1 of lead solution at 2 mV min-1. The detection limit and sensitivity for the proposed method were improved by 26 times and 8.8 times than those of HGAAS��respectively. The detection limit (3s) of the proposed method for lead in real sample was 0.96 ng L-1 for taking 5.0g sample at a 2 mVmin-1 sensitivity range setting. The method was applied to the determination of lead in seasoning samples, such as monosodium, glutamate, white sugar, table salt and vinegar. The recoveries of spiked Pb from real samples are in the range of 90.8-107% for real samples. ACKNOWLEDGEMENTS The authors express thanks to the Natural Science Foundation of Hebei Province and the Specialized Research Funds of China Education Ministry for much support to the studied subjects (No.203110) ��No.20050075003). REFERENCES
[1] Ojeda C. Bosch, Rojas F. Sanchez, Pavon J. M et al., Talanta, 1995, 42: 1195.
[2] EL-SAYED A. A Y, EL-SALEM N A. Anal. Sci., 2005, 21: 595.
[3] Yan Z, Sun H W, Zhang J S. Spectroscopy and Spectral Analysis, 1994, 14(5): 63.
[4] Wang W X, Sun H W. Physics Test and Chemical Analysis (Part B: Chemical Analysis), 2001, 37(1): 27.
[5]Sun H W,Li L Q. Journal of the Iranian Chemical Society, 2005, 2, (4): 268.
[6] Sun H W. Spectroscopy and Spectral Analysis, 2003, 23: 386.
[7] Sun H W. Atomic spectroscopy and trace Analysis��Hebei University Pres, Baoding, 2001.
[8] Sun H W. Atomic spectrometry, High education Pres, Beijing, 2002.
[9] Sun H W. Journal of Hebei University, 2001, 21(3): 256.
[10] Sun H W, Ha J. Sun J M et al., Fresenius' J. Anal. Chem., 2001, 371: 1154.
[11] Sun H W, Ha J, Sun J M et al., Anal. Bioanal. Chem., 2002, 374: 526.
[12] Yi J H, Chen T S. Environmental Monitoring in China, 1998, 14 (6): 30.
[13] Hu Y S, Su W Z. Journal of analytical Chemistry, 1996, 24: 371.
[14]Zhao Z H, Tian D H, Quan W Z. Environ. Sci., 1995, 16(4): 54
[15] Hashem Elham Y. Spectrochim. Acta, A 2002, 58: 1401.
[16]Chen Z G, Chen Y. Chinese Journal of analytical Chemistry, 1999, 27: 862.
[17] Zhang D Q, Yang L L, Sun H W. Anal. Chim. Acta, 1999, 395: 173.
[18] Ha J, Sun H W, Sun J. M et al., Anal. Chim. Acta, 2001, 448: 145.
[19] Sun H W, Zhang D Q, Ha J et al., Anal. Sci., 2002, 18: 603.
[20] Chai Z F. Environ. Sci., 1978, 3: 47.
[21] Chai Z F, Zhu H M. Introduction to Trace Element Chemistry, Atomic Energy Press, Peking, China, 1994, 201.
[22] Kobayashi R, Imaizumi K. Anal. Sci., 1991, 6: 83.
[23] Kobayashi R, Imaizumi K. Anal. Sci., 1991, 7: 447.
[24] Stiddik Z H, Newman R A. Anal. Biochem., 1988, 172: 190.
[25] Lewande O. Am. Diet. Assoc., 1991, 91: 1572.
[26]Jiang D Z, Li y L, Hu Y H. Chinese Journal of Health Laboratory Technology, 2005; 15: 563.
[27] Gomez M M, Gasparic T, Plascios M A et al., Anal. Chim. Acta, 1998, 374: 241.
[28] Siemer D D, Hageman L. Anal. Chem.. 1980, 52: 105.
[29] Lee S H, Jung K H, Lee D S. Talanta, 1989, 36: 999.
[30] Hladky Z, Risova J, Fisera M. J. Anal. At. Spectrom., 1990, 5: 691.
[31] Baxter D C, Frech W. Anal. Chim. Acta, 1989, 225; 175.
[32]Zhou T. Modern Preventive Medicine, 2005, 32; 790.
[33] Zhang D Q, Yang L L, Sun H W. Achievement and Challenge of Analytical Chemistry��Southwest Normal School Press, Congqing, 2000.
[34] Zhang B G, Wang Y, Wang X S et al., Talanta, 1995, 42: 1095.
[35] Cabon J Y, Madec C L. Anal. Chim. Acta, 2004, 504: 209.
[36] Sun H W, Ha J, Sun J M. Chemical Journal on Internet, 2003, 5(3): 26.
[37] Sun H W, Liang S X, Ha J et al., Chemical Journal on Internet, 2003, 5(3): 23.
[38] Xie L H. Physics Test and Chemical Analysis (Part B: Chemical Analysis) 2005, 41(5): 334.
[39] Song J Q. Physics Test and Chemical Analysis (Part B: Chemical Analysis) 2004, 40 (3): 172.
[40] Liang S X, Ha J, Sun H W et al.��Chem. Analysis, 2003, 48: 723. ������������ԭ�����չ��������ں��������е�Ӧ��
�ﺺ�� ������ ������
(�ӱ���ѧ��ѧ�뻷����ѧѧԺ���ӱ�ʡ������ѧ�����ص�ʵ���ң�071002, ����)
ժҪ �������ӹ����ǻ��ڲ����ź�ǿ���沨���ı仯�ʡ������������һ�����͵ĵ�����������ԭ�����շ��ǻ��ڲ����ź�ǿ����ʱ��ı仯�ʡ����Ľ�����������ԭ�����պ��⻯��ԭ�����ճ����ź�ģ�ͼ��䵼���źŷ��̡������˵�����������ԭ�����չ�����ԭ�������ԡ������ź�ǿ���������Ũ��֮������õ����Թ�ϵ���뵼�����ӹ�����������������ԭ�����չ�����ȣ�������������ԭ�����չ������нϸߵ������Ⱥͽϵ͵ļ���ޡ������˵�����������ԭ�����չ����ں��������е�Ӧ�á�
�ؼ��� �⻯�ԭ�����չ��ף��ź�ģ�ͣ���������ԭ������������
��