| http://www.chemistrymag.org/cji/2006/089059pe.htm | Sep. 8,
2006 Vol.8 No.9 P.59 Copyright |
Miao Mingming 1,2, Huang Yun 1,2,
Kong Weishong 1, Chen Zhangyu 1, Yang Guangyu 1,2*
(1 Chemical Laboratory; Yunnan
Academy of Tobacco Sciences, Kunming 650106; 2 Technology Center of Hongta Group, Yuxi 653100)
Abstract A new method for the
determination of mercury and selenium in tobacco by inductively coupled plasma mass
spectrometry (ICP-MS) using isotope dilution calibration was studied. The samples were
digested with aqua regia in a microwave oven. The isotope ratios used for
quantification were 201Hg/202Hg and 77Se/82Se.
A NaBH4 solution was used as reducing agent. Five certified tobacco samples
were analyzed and the concentrations obtained were in good agreement with the certified
values. The detection limits in the samples were 0.5 ng g-1 for mercury and 2
ng g-1 for selenium, respectively. The method is precise, accurate and rapid.
Keywords mercury; selenium; ICP-MS; isotopic dilution calibration; microwave
digestion; Tobacco
1. INTRODUCTION
Mercury and selenium are important metal ions existing in tobacco. The Quality
Standards says that the concentration of mercury should not be exceeding 0.2 m g g-1
in tobacco because of its high toxicity. However, the selenium is helpful to
our healthy when its concentration in tobacco within an appropriate range. [1] Therefore,
the determination of trace mercury in tobacco and tobacco additive is important.
Presently, there are several analytical methods for the determination of mercury and
selenium including spectrophotometry, atomic absorption spectrometry, fluorescence and
mass spectrometry (MS). [2-18] Among the analytical techniques employed,
inductively coupled plasma mass spectrometry (ICP-MS) is especially important due to its
several advantages, such as its multi-element capacity, high sensitivity and ability to
measure isotopic ratios, which allows isotope dilution calibration (ID).[11-18]
In this work, a method for the determination of mercury and selenium in tobacco, after
microwave-assisted acid digestion with aqua regia of the samples followed by
chemical vapor generation with retention of the analyte vapor in an Ir-treated graphite
tube of an electrothermal vaporizer using isotope dilution calibration (ID-CVG-ETVICP-MS)
is optimized and described. The results show that this method is precise, accurate and
rapid.
2 EXPERIMENTAL
2.1 Instrumental
All the measurements were carried out on an inductively coupled plasma mass
spectrometer ELAN 6000 (Perkin Elmer SCIEX, Thornhill, ON, Canada). For the ETV system, a
hydride generator MHS-15 (Perkin Elmer, CO, USA) was coupled to a Perkin-Elmer HGA 600 MS
electrothermal vaporizer and a Perkin Elmer AS-60 autosampler and manually operated, as
described previously.18 A 3% (m/v) sodium borohydride solution stabilized with
1% (m/v) sodium hydroxide was used as reducing agent in the MHS-15. The reducing agent was
injected during 5 s using an argon pressure of 250 kPa. The generated vapors were
transported to the ETV by an Ar flow through a 10 cm glass tube (0.1 cm i.d.) connected to
a polytetrafluoroethylene (PTFE) tube (60 cm long, 0.5 cm i.d.). Pyrolytic coated graphite
tubes (Perkin-Elmer, Part No. B050 8371) were used. Argon of 99.996% purity (Kunming
Yangqi, Kunming, P.R.China) was used. An internal Ar flow rate of 0.1 L min-1
in the vaporizer, optimized previously,[18] summed to the "nebulizer" gas flow rate
of 1.06 L min-1 during vaporization, resulting in a total flow rate of 1.16 L
min-1, transported the aerosol to the plasma. The instrumental conditions are
shown in Table 1 and the ETV temperature program, which also was optimized previously[18]
is shown in Table 2. The samples for the microwave digestion were weighed using a M2P
microbalance (Sartorius, Göttingen, Germany). A Model WL 5001 microwave system (1000
W, Fei Yue Analytical Instrument Factory, Shanghai, China) was employed for the digestions
of the samples.
2.2 Reagents and materials
All the reagents were of analytical grade. The water used was de-ionized in a Milli-Q
system (Millipore, Bedford, MA, USA). Nitric acid (High purity grade, Tianjing, P.R.China)
and hydrochloric acid (High purity grade, Tianjing, P.R.China) were used. The reducing
agent was prepared by dissolving NaBH4 (Fluka, Buchs, Switzerland) in sodium
hydroxide (High purity grade, Tianjing, P.R.China) stored in a polyethylene flask and kept
under refrigeration for no longer than two days. A stock solution of IrCl3
(Fluka, Buchs, Switzerland), 1000 mg L-1 Ir, was used for the treatment of the
tube with the permanent modifier as described previously.20 The enriched
isotope materials were from the Cambridge Isotope Laboratories Inc. (Andover, MA, USA).
The abundances of the enriched isotopes were: 96.35% of 201Hg and 93.48% of 77Se.
The isotope compositions of the enriched materials were measured and the found values
agreed with the informed values. Stock solutions of 30 mg L-1 for mercury and
200 m g L-1 for selenium were prepared by dissolution of an accurately weighed
amount of the solid material (HgO and elemental Se) in nitric acid and diluted in 5% v/v
HNO3. The following isotope ratios were used in the calculation of the
concentrations: 201Hg/202Hg and 77Se/82Se. The
ratio of the signal intensities of the isotopes in the samples without the addition of the
enriched isotope was compared to the natural ratio in order to check for spectral
interference and mass discrimination. The correction factor for the mass discrimination
was calculated by comparing the natural isotope ratio with the measured ratio for the
sample without the spike. The software automatically uses the correction factor to correct
the measured altered ratio.
2.3 Sample preparation
The samples analyzed are Certified Chinese Standard Material. The standard values were
determined by Atomic Absorption Spectrum (AAS), Atomic Fluorescence Spectrum (AFS),
Neutron Activation Analysis (NAA), Inductively Coupled Plasma-Mass Spectrometry (ICP-MS),
and Ion Chromatography (IC) method.
An aliquot of approximately 250 mg of the sample was weighed directly
in a PTFE flask and put in the microwave oven. Then, the solutions of the materials
enriched with the isotopes 201Hg and 77Se were added to the flask in
an adequate amount in order to obtain an altered ratio close to 1, to minimize the
measurement errors. Due to the low concentrations of selenium and mercury in the samples,
masses of the enriched materials at ng level were added. The masses were calculated using
the ID equation for an altered ratio of 1 and converting the masses to volumes of the
enriched materials solutions. The sample aliquot without spike was digested with 3.5 mL aqua
regia plus 1.0 mL of deionized water in the microwave bombs. The sample aliquot with
the spikes was also digested with 3.5 mL aqua regia plus a certain volume of
deionized water, which was the complement to 1 mL of the volumes of the spiking solutions.
In this way, all sample aliquots, without and with spikes, were digested in the same
volume of liquid. The bombs were sealed tightly and then positioned in the carousel of the
microwave oven. The system was operated at full power for 6.0 min. After the digestion was
finished, all of the solutions were colorless, indicating an effective digestion.
Following that, 1 mL of the digested sample was mixed with 1 mL of concentrated
hydrochloric acid and heated to 90 °C for 30 min, in order to guarantee the lower
oxidation state for selenium, favoring its hydride generation.[7] After cooling
the solution, the final volume of 10 mL was made up with deionized water, which was not
strictly necessary, as isotopic dilution calibration was used, assuming that equilibration
of the added isotope with the isotope in the sample have occurred during digestion.
However, this final dilution to the same final volume for each sample aliquot, allowed a
better control of the counting signals.
2.4 Analytical procedure
For the determination of mercury and selenium in samples, a 1 mL aliquot of the final
sample solution (without or with spike), was transferred to the reaction flask of the
hydride generator and the temperature program of the ETV was started. The glass tube of
the hydride generator was manually introduced into the graphite tube. During the
pre-heating step, the reducing agent was added to the reaction flask for 5 s and the
generated vapors were transferred to the graphite tube by using argon for 30 s in the
MHS-15. Before the vaporization step, the glass tube was removed and the graphite tip of
the ETV arm closed the dosing hole of the graphite tube. Reading was carried out during
the vaporization stage, in which the vapor is transported from the ETV to the plasma by a
total argon flow rate of 1.16 L min-1.
3 RESULTS AND DISCUSSION
3.1 Sample digestion
The chemical vapor generation conditions and the
electrothermal vaporizer temperature program were optimized previously for the
determination of mercury and selenium of 3.5 mL of aqua regia used. Lower volumes
do not lead to accurate results neither for mercury nor for selenium, and other analytes
in sediments, as slurries in an aqua regia plus HF medium, using external [18]
or isotopic dilution calibration.[19] The isotopes 202Hg (natural
abundance of 29.8%) and 82Se (natural abundance of 8.73%) were chosen as
reference isotopes, taking into account the absence of spectral interference and their
natural abundances. In this way, the concentrations were obtained by measuring
simultaneously the following altered isotope ratios: 201Hg/202Hg and
77Se/82Se.
3.2 Effect of the hydrochloric acid treatment and the aqua regia concentration
In order to generate the vapor, the sample should be completely digested, meaning that all
organic compounds containing the analytes should be destroyed. The microwave-assisted
digestion of the tobacco sample with aqua regia efficiently digested the tobacco
samples, as clear solutions were obtained. As already mentioned, the isotope dilution is
an ideal internal standardization, as the internal standard, in this case, is one isotope
of the same element. In principle, if a reasonable fraction of the selenium, after the
digestion, was present in its lower oxidation state, which is able to generate the
hydride, the isotope calibration should compensate for the fraction of the selenium, which
is on its higher oxidation state and does not generate the hydride.[6] In this
way, a procedure was attempted, in which the sample solution, after digestion, was not
heated with hydrochloric acid, but was only taken to a final volume of 10 mL with 1 mol L-1
hydrochloric acid. The obtained concentrations for selenium were significantly higher
(from 19% to 50%) than the certified values, indicating that the heating with hydrochloric
acid, which converts Se(VI) to Se(IV) is required even when isotope dilution calibration
is employed, what could indicate that the Se(IV) fractions in the spike and in the sample
are different. However, for mercury, the obtained results, with and without heating with
hydrochloric acid, are in agreement with the certified values according to the t-test
for a 95% confidence level. Thus, 1 mL of the sample solution was mixed with 1 mL of
hydrochloric acid and heated to 90
The aqua regia volume added to the sample mass of around 250 mg for the microwave-assisted digestion was investigated, using the GB-2102 sample. A volume of 3.5 mL of aqua regia was used. Lower volumes do not lead to accurate results neither for mercury nor for selenium, being the obtained concentrations much lower than the certified ones, indicating that part of the analytes are not in the appropriate forms to generate the cold vapor of mercury or the hydride of selenium. Most probably, the lower volume is not enough to mineralize completely the sample.
Table 1 ETV-ICP-MS operational parameters
| Parameters | |
| RF power (W) | 1000 |
| Gas flow rate | (L min-1) |
| Principal | 15 |
| Intermediate | 1.2 |
| Carrier | 1.06 |
| Sampler and skimmer cones | Pt |
| Dwell time (ms) | 25 |
| Sweeps per reading | 1 |
| Reading per replicate | 150 |
| Resolution at 10% of the peak height | 0.7 u.m.a |
| Auto lens | On |
| Signal measurement | Peak area |
Step |
Temperature |
Ramp(s) |
Hold(s) |
Gas flow |
Cooling |
20 |
1 |
5 |
300 |
Pre-heatinga |
150 |
5 |
0 |
0 |
Pre-heatingb |
150 |
1 |
30 |
0 |
Cooling |
20 |
1 |
8 |
0 |
Vaporizationc |
2000 |
1 |
20 |
100 |
Cleaning |
2200 |
5 |
5 |
300 |
Cooling |
20 |
1 |
5 |
300 |
a glass tube introduction into the graphite tube; b analyte vapor collections on the graphite tube; c vaporization and reading. |
||||
3.3 Figures of merit and analytical
application
The limit of detection (LOD) is defined as the minimum concentration or weight of analyte
that can be detected at a known confidence level. The limits of detection, in the sample,
calculated as a function of the enrichment of the isotopic spike and the linear
calibration detection limits for each isotope 21 are 0.5 and 2 ng g-1
for mercury and selenium, respectively, indicating that the proposed procedure is able to
detect very low concentrations of the analytes in tobacco samples. In a very recent paper,
Santos et al.5 have investigated the chemical vapor generation for
mercury and selenium in biological samples as slurries in different media, prior to
detection by in ICP-OES, using external calibration. For 20 mg of solid sample in 15 mL of
slurry, the obtained detection limits (3 s, n = 10) for the proposed procedure were 80 and
100 ng g-1 for mercury and selenium, respectively, much higher (about 110 times
for mercury and 33 times for selenium) than those obtained in this work, as expected due
to the higher sensitivity of the ETV-ICP-MS technique. Vieira et al.18
have proposed a method for the determination of arsenic, mercury, selenium and tin in
sediment by slurry sampling CVG-ETV-ICP-MS, also with trapping of the vapor on a treated
graphite tube, but using slurry sampling and external calibration against aqueous
standards, obtaining about the same LOD (3 s, n = 10) for mercury, 0.80 ng g-1,
as in this work, and 300 times better LOD for selenium, 0.01 ng g-1.
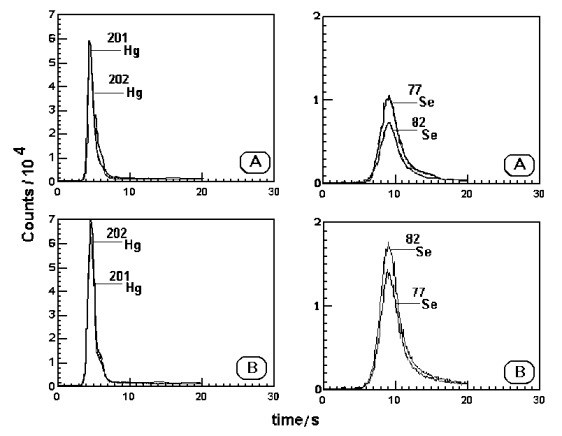
Figure 1. Transient signals for Hg and Se in reference material GB-2102 (a) without and (b) with the addition of the enriched
isotope.
In Figure 1 some typical
transient signals for mercury and selenium in reference material GB-2102, before and after
addition the enriched isotopes, are shown. As shown in the Figure the condition of an
altered isotopic ratio close to 1 was obtained for mercury and selenium.
The accuracies of the procedures were estimated by the analysis of fix
certified reference materials. As shown in Table 3, the found concentrations are in
agreement with the certified values, according to the t-test for a confidence level
of 95%. The relative standard deviations (RSD) were the range from 2.8 to 3.4% for
selenium and from 2.8 to 4.1%, for mercury, indicating an adequate precision. Probably, an
efficient digestion of the sample, with complete destruction of the analytes organic
compounds by microwave digestion with aqua regia was attained, providing the
equilibration of the added isotopes with the isotopes in the sample, which is a basic
requirement for the ID calibration. After equilibration, there is no need to use an exact
volume of the sample solution.
Table 3 Determined concentrations in m
g g-1 for mercury and selenium in certified tobacco samples by CVG-ETV-ICP-MS
using isotope dilution calibration after microwave digestion with aqua regia and
treatment with hydrochloric acid to reduce the oxidation number of selenium (n = 5)
Samples |
Certified (ng/g) |
Found (ng/g) |
RSD% (n = 5) |
|||
Hg |
Se |
Hg |
Se |
Hg |
Se |
|
GB-2102 |
22.8 |
68.5 |
20.5 |
69.8 |
3.4 |
3.0 |
GB-2108 |
18.2 |
76.8 |
16.8 |
78.4 |
3.6 |
2.8 |
GB-2124 |
56.4 |
87.5 |
58.2 |
85.4 |
2.8 |
3.2 |
GB-2180 |
68.9 |
84.8 |
65.8 |
86.9 |
4.1 |
2.8 |
GB-2194 |
35.6 |
61.2 |
38.2 |
59.4 |
3.9 |
3.4 |
4 CONCLUSIONS
It was demonstrated that mercury and selenium can be determined in tobacco samples
after microwave-assisted acid digestion with aqua regia followed by their
determination by CVG-ETVICP-MS using vapor trapping in the graphite tube and isotopic
dilution calibration. The treated tube with Ir was efficient for the trapping of the
mercury cold vapor and the selenium hydride. The proposed procedure using ID compensate
eventual analyte loss during the steps of sample preparation, including digestion and
reduction of the oxidation state for selenium. The reduction of the oxidation state for
selenium with hydrochloric acid prior to its hydride generation is necessary, even using
isotope dilution calibration. For mercury, the reduction step with hydrochloric acid can
be omitted.
Acknowledgement This work was supported by the National Natural Science Foundation of China (20471051) and the Key Natural Science Foundation of Yunnan Province (2005K021)
REFERENCES
[1] Chen Z Y, Kong WS, Liu W et al. Chinese. J. Tob. Res. 2003, 2, 115
[2] Matusiewicz, H.; Sturgeon, R. E.; Berman, S. S.; J. Anal. At. Spectrom. 1989, 4, 323.
[3] Vieira, M. A.; Ribeiro, A. S.; Curtius, A. J.; Anal. Bioanal. Chem. 2004, 380, 570.
[4] Cal-Prieto M J, Felipe-Sotelo M, Carlosena A et al. Talanta 2002, 56, 1.
[5] Dos Santos, E. J.; Herrmann, A. B.; Frescura, V. L. A.; Curtius, A. J.; Anal. Chim.
Acta 2005, 548, 166.
[6] Welz, B.; Sperling, M.; Atomic Absorption Spectrometry, 3rd ed., Wiley-VCH:
Weinheim, 1999.
[7] Petersson, J.; Olin, A.; Talanta 1991, 38, 413.
[8] Jin, Q.; Liang, F.; Zhang, H.; Zhao, L.; Huan, Y.; Song, D.; Trends Anal. Chem. 1999,
16, 479.
[9] Brisbin, J. A.; Caruso, J. A.; Analyst 2002, 127, 921.
[10] Houk, R. S.; Anal. Chem. 1986, 58, 97A.
[11] Dias, L. F.; Saint’Pierre, T. D.; Maia, S. M.et
al. Spectrochim. Acta, Part B 2002, 57, 2003.
[12] Pozebon, D.; Dressler, V. L.; Curtius, A. J.; J. Anal. At. Spectrom. 1998, 13, 1101.
[13] Saint' Pierre, T. D.; Maranh?o, T. A.; Frescura,
V. L. A.; Curtius, A. J.; Spectrochim. Acta, Part B 2005, 60, 605.
[14] Vanhaecke, F.; Boonen, S.; Moens, L.; Dams, R.; J. Anal. At. Spectrom. 1997, 12, 125.
[15] Tibi, M.; Heumann, K. G.; J. Anal. At. Spectrom. 2003, 18, 1076.
[16] Chang, C.; Jiang, S.; J. Anal. At. Spectrom. 1997, 12, 75.
[17] Maia, S. M.; Pozebon, D.; Curtius, A. J.; J. Anal. At. Spectrom. 2003, 18, 330.
[18] Vieira, M. A.; Saint' Pierre, T. D.; Welz, B.;
Curtius, A. J.; J. Anal. At. Spectrom. 2004, 19, 297.
[19] Vieira, M. A.; Ribeiro, A. S.; Dias, L. F.; Curtius, A. J.; Spectrochim. Acta, Part B
2005, 60, 643.
[20] Vieira, M. A.; Curtius, A. J.; Welz, B.; Spectrochim. Acta, Part B 2002, 57, 2057.
[21] Yu, L. L.; Fassett, J. D.; Guthrie, W. F.; Anal. Chem. 2002, 74, 3887.
缪明明 1,2, 黄 云 1,2, 孔维松 1, 陈章玉1,杨光宇 1,2
(1云南烟草科学研究院化学室,昆明 650106;2红塔烟草集团公司技术中心,玉溪 653100)
摘要 研究了用同位素稀释,电感耦合等离子体质谱(ICP-MS)法测定烟草中的硒、汞,样品用王水微波消化,定量的同位素比例为 201Hg/202Hg 和 77Se/82Se,用NaBH4作还原剂,用该法发测定了五个烟草行业标样,方法的检测限为:汞 0.5 ng g-1 和硒 2.0 ng g-1,该方法灵敏度高,快速、准确,适合于烟草中痕量硒和汞的测定。
关键词 汞;硒;电感耦合等离子体质谱;同位素稀释;微波消化;烟草