| http://www.chemistrymag.org/cji/2006/08b067pc.htm | Nov. 1,
2006 Vol.8 No11 P.67 Copyright |
Xiang Xiaolan1,2 Yu Bin2 He Jinlan1
(1Department of Chemistry, Zhanjiang Normal
College, Zhanjiang 524048; 2College of Sciences, Nanjing University of
Technology, Nanjing 21000)
Keywords micellar electrokinetic capillary chromatography, high frequency conductivity detection, amino acid, medicine analysis
胶束电动毛细管色谱-高频电导检测定量分析药物中的氨基酸
项小兰
1,2 俞斌 2 何金兰1(1湛江师范学院化学系,广东 524048;2南京工业大学理学院,南京 210009)
2006年9月26日收稿;广东省自然科学基金资助项目 (粤科基办[2005]04号)
摘要
首次采用胶束电动毛细管色谱-高频电导检测定量分析了药物中的氨基酸。在酸性条件下(pH3.8),1mmol/L的十二烷基硫酸钠(SDS)能有效地克服毛细管壁对带正电荷的氨基 酸的吸附;在磷酸二氢钠的缓冲体系中,添加5mmol/L的甲酸,6分钟内完成5种氨基酸的分离检测;分离效率在0.72×105~ 2.48×105N/m之间,检出限在0.03-2.01mmol/L之间。全文对高频电导检测机理及影响因素进行了详细的探讨。关键词 胶束电动毛细管色谱,高频电导检测,氨基酸,药物分析 自1984年Terabe首次用1nmol/L的十二烷基硫酸钠作为胶束,用毛细管电泳成功分离儿茶酚后[1],胶束电动毛细管色谱广泛地用于亲水和疏水物质的分离[2-4]。药物中氨基酸(Amino acids, AAs)分析的主要问题是:多数AAs既无光活性又无电活性;常常需要进行光化学衍生[5,6]或电化学衍生[7]后,才能进行检测。化学衍生不仅烦琐费时;而且,污染样品,带来误差。
毛细管电泳高频电导检测是一种非接触式电导检测[8,9],检测电极与溶液不直接接触;检测电极之间一般施加20~250kHz的高频电压,分离过程中,记录两检测电极之间的高频电流的变化;当被分离的组分流经两检测电极的间隙时,由于分离组分与缓冲体系组分的电导不同,至使高频电流发生改变,从而被分离组分得到检测。显然,被检测组分与分离缓冲体系之间的电导差异愈大,高频电流的变化愈大,检测灵敏度愈高。
氨基酸在其等电点以外的溶液中均带电荷,非常适于采用这种非接触式电导检测方式[9]。本文以十二烷基硫酸钠作为胶束,详细讨论了胶束电动毛细管色谱中,影响分离度和高频电导检测灵敏度的因素;在优化的条件下,在正常极性条件下,6分钟内完成了5种氨基酸的快速分离,分离效率在0.72×105-2.48×105N/m,检出限在0.03-2.01mmol/L之间;并将方法用于药物中的氨基酸分析。
1 实验部分
SYS/CE 99高频电导检测器-高效毛细管电泳仪(中山医科大学科技开发部)。未涂层毛细管柱(ID.100mm,OD.360mm,总长60cm 有效长度54cm,河北省永年锐丰色谱器件有限公司)。
十二烷基磺酸钠(SDS,广州化学试剂玻璃仪器公司);磷酸二氢钠(汕头光华试剂厂);甲酸、乙酸、乳酸(天津科密欧化学试剂开发中心);以上试剂均为分析纯。苯丙氨酸(Phe),精氨酸(Arg),丙氨酸(Ala),谷氨酸(Glu)和天冬氨酸(Asp)均为Sigma公司色谱纯。实验用水为反渗透去离子水。
1.2 实验方法
分离电压20.5kV,进样高度15 cm,进样时间10s,阴极检测,分离温度25℃。所有溶液在进入毛细管之前,经0.45μm微孔滤膜过滤;每次电泳前用分离缓冲溶液冲洗1min。高频电导检测器的输出信号经数据工作站采集到微机中进行实时数据处理、图形显示和数据文件存储。
1.3 样品溶液的制备
取复方氨基酸注射液(广州绿十字药业有限公司)10ml各三份,其中一份加待测氨基酸各10mg作为回收测试样,另二份作为平行测试样;经045μm微孔滤膜滤过,然后减压浓缩至0.5ml,待测。
2
结果与讨论2.1 SDS浓度的影响
一般情况下,熔硅毛细管壁带负电荷;在酸性条件下,多数氨基酸带正电荷。那么,在酸性条件下分离,多数氨基酸容易被管壁吸附;正常极性下检测时,分离峰拖尾、分离度下降。为了克服吸附效应,我们在负极检测条件下,添加十二烷基硫酸钠(SDS)作为胶束,有效克服了管壁吸附问题。
但是,非接触式的高频电导检测实际上是一种间接检测方式,背景电解质的浓度不仅影响分离效率,还直接影响检测灵敏度。图1是在同一酸度(pH3.8)下,不同浓度SDS对氨基酸的分离效率和检测灵敏度的影响。
SDS从1mmol/L增加至5mmol/L(图1B,C,D),样品峰高从200mV降至100mV左右。我们知道,高频电导检测记录的信号是高频感应区带(即,检测窗口)的电流,经计算机模/数转换后以mV的形式记录。当背景电解质与样品区带的电导差异愈大时,两者的电流差异也愈大。SDS浓度的增加,背景电解质的电导增加,其电流逐渐增加;背景电解质和样品区带之间的电流差异逐渐减少;所以,检测灵敏度下降。
同时,SDS浓度增加使分离电流增大,使信噪比过大,分离效率明显下降(图1D)。而图1A中SDS的浓度为零,带正荷的氨基酸被管壁吸附,分离效率更差。可见,1mmol/LSDS存在于缓冲体系中能克服管壁的吸附效应,分离效率明显改善。
2.2 缓冲体系组分与浓度的影响
在酸条件下,我们选择了乙酸~乙酸钠、磷酸二氢钠、邻苯二甲酸氢钾、2-3,5-二硝基水杨酸等作为缓冲体系组分,在不同浓度下分别与1mmol/L的SDS组成分离缓冲体系。发现乙酸~乙酸钠体系基线稳定,噪声小,但出峰慢,峰展宽严重;邻苯二甲酸氢钾和2-3,5-二硝基水杨酸体系电导率比较大,分离电流大,基线不稳定;磷酸二氢钠作为缓冲体系,其浓度在5~10mmol/L时,都能获得稳定的基线,而且其电导率比较低,分子量比较小,对氨基酸的检测灵敏度比较高。综合分离时间、分离容量和灵敏度的考虑,我们选择7.5mmol/L的磷酸二氢钠作为缓冲体系组分。
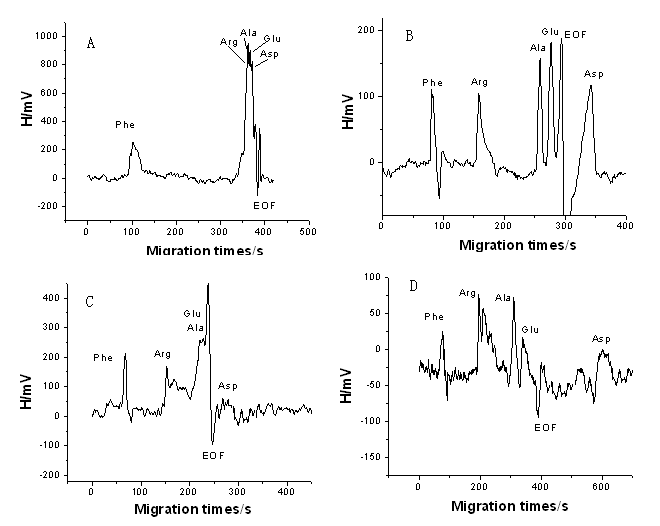
图1 SDS浓度对氨基酸分离和检测的影响
Fig.1 Effect of the conc. of SDS for separation and detection
Separation condition:in 7.5mmol/L sodium dihydrogen phosphat,5 mmol/L formic acid,pH3.8
A)with 0mmol/LSDS,B)with 1mmol/LSDS,C)with 3mmol/LSDS,D)with 5mmol/LSDS 2.3 有机添加剂的影响
在1mmol/LSDS、7.5mmol/L的磷酸二氢钠(pH3.8)的溶液中,分别添加甲酸、乙酸和乳酸组成三种不同缓冲体系。三种有机酸的最佳添加量分别为5、8和7.5mmol/L。在优化条件下,从甲酸、乙酸到乳酸随着分子量的增加,三体系的检测灵敏度和分离效率基本上依次呈下降趋势(见图2)。有机添加剂一般能改善体系的分离效率,但是在本研究中,甲酸的添加比乙酸和乳酸的添加的灵敏度更高,这主要是对间接检测方法的影响。
根据间接检测法的浓度检出限CD的计算公式:
式中,Cb为背景电解质的浓度;TR为单位分析物置换背景电解质的分子数,即,转换比;DR为背景电解质的信噪比,又称为动态储备。
甲酸存在于背景电解质中,使公式(1)中TR值比乙酸和乳酸的体系大,即样品分子的转换比TR值随着背景电解质的分子量的减小而增加;TR值增加,检出限下降,所以灵敏度提高。
分离效率随甲酸、乙酸、乳酸的分子量的增加而下降,则是因为组分的迁移时间随着缓冲体系的浓度的增加而增加,分离峰展宽,分离效率下降。可见,间接检测模式中,适当的有机添加剂不仅可以改善分离效率,还能有效地提高单个样品分子的转换比,从而提高检测灵敏度。
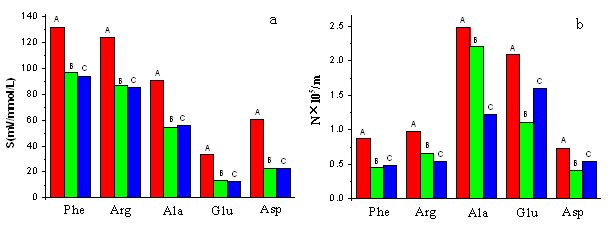
图2 有机添加剂对氨基酸分离和检测的影响 a)灵敏度;b)分离效率
Fig.2 Effect of organic reagent on separation and detection
a)sensitivity,b)separation efficiency
Running buffer:in 1mmol/LSDS,7.5mmol/L sodium dihydrogen phosphat (pH3.8),
A:with 5 mmol/L formic acid,B:with 8 mmol/L acetic acid,C:with 8 mmol/L lactic acid.
2.4
定量分析特征参数、分离效率及样品分析结果图3B为优化体系:1mmol/LSDS+7.5mmol/L sodium dihydrogen phosphat+5 mmol/L formic acid (pH3.8)20.5 kV下,5种标准氨基酸的电泳分离图(3A)及复方氨基酸注射液电泳分离图(3B)。在此条件下,用峰高-浓度绘制工作曲线。校正曲线特征参数及各组分的分离效率列在表1;按1.3制备的样品测定结果及加标回收率列在表2。表2可知,测定结果与证书值一致。
表1 定量分析特征参数及各组分的分离效率
| 组 分 Component |
检出限 Detection limit (mmol/L) (k=3,n=11) |
线性范围 Linear range (mmol/L) |
回归系数 Linear regression |
相对标准偏差* RSD(%), (n=10) |
分离效率 Separation efficiency (N/m) |
||
| tm | h | (c ) | |||||
| Phe Arg Ala Glu Asp |
0.03 0.63 0.74 2.01 0.92 |
0.2-2.0 0.2-5.0 0.2-10.0 1.0-9.0 1.0-8.5 |
0.999 0.999 0.999 0.999 0.999 |
1.37 2.38 2.28 4.55 3.91 |
2.78 1.98 2.05 1.58 1.16 |
(1.1) (2.5) (5.0) (4.5) (4.0) |
0.88×105 0.97×105 2.48×105 2.09×105 0.72×105 |
c 精度测定浓度(the measurement conc. for RSD)(mmol/L)
表2 氨基酸注射液分析结果
Table 2 Determination results for the injection of amino acids
| 组分 Component |
样品1 Sample 1 |
样品2 Sample 2 |
回收率(%) Recovery rates |
证书值 certificate |
| Phe Arg Ala Glu Asp |
31.0 25.0 22.0 712.0 22.0 |
33.0 25.0 21.0 712.0 21.0 |
97.8 95.2 97.3 93.3 95.5 |
30.0 23.0 20.0 750.0 20.0 |
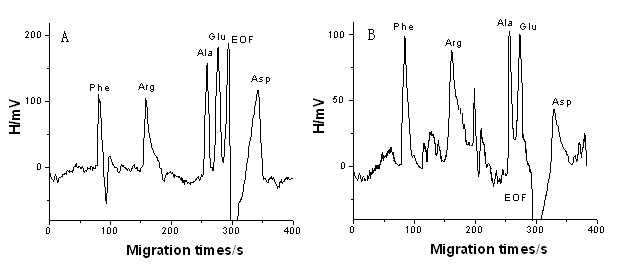
图3 优化条件下氨基酸标准和样品的毛细管电泳图
Fig.3 Electrophorograms of amino acid standards(A)and sample(B) in optimum condition
Running buffer:1mmol/LSDS,7.5mmol/L sodium dihydrogen phosphat,and 5 mmol/L formic acid (pH3.8)
Running voltage:20.5 kV
3 结论
2)在高频电导检测中,高频电压不变的条件下,缓冲溶液与样品区带之间的电流差异愈大,检测灵敏度愈高。增加缓冲溶液与样品区带的浓度差异或者增加两者摩尔电导率的差异,都可以导致电流差异增加。显然,增加样品的浓度来提高灵敏度是没有意义的,应该尽量降低背景电解质的浓度。但是,背景电解质的浓度太低,导致分离容量下降;所以,增加两者的浓度差异是有限的。
那么,选择与样品摩尔电导率差异大的背景电解质做缓冲体系是提高非接触式高频电导检测灵敏度主要途径。实际上,高频电导检测也是一种间接检测,即,利用背景与样品区带的信号差异进行样品识别。在一般的间接检测技术中,样品信号为零,要改善检出限,多采用高背景信号的缓冲体系就可以获得高的转换比[4,10,11]。但是,在高频电导检测中,样品信号不是零,样品自身是具有电导率的,高背景信号的缓冲体系完全不适合非接触式高频电导检测技术。实践表明,采用适当的低电荷、低分子量的组分作缓冲体系对改善方法检出限和提高分离效率两个方面都是有效的。 REFERENCES
[1] S Terabe, K Otsuka, K Ichikawa, et al. Anal. chem., 1984,56:111-122.
[2] 邓宁,何友昭,苏庆德,分析化学,2005,33(10): 259-262.
[3] 卢小玲,倪坤仪,屠洁等,分析化学,2006,34(2):259-262.
[4] 徐远金,许桂苹,魏远安,色谱,2006,24(1): 135-38.
[5] R M Lartorre, J Saurina, S H Cassou, J. Chromatogr., A 2002,976:55-64.
[6] 陈义,卢小宁. 高等学校化学学报, 2002,23(5):822-824.
[7] Weng Q. F., Jin W. R., Anal. Chim. Acta, 2003,478:199-207.
[8] 徐健君,翟海云,陈缵光等. 应用化学, 2005,22(6):581-585.
[9] 陈缵光,莫金垣,等学校化学学报, 2002, 23(5):801-804.
[10] T Takeuchi, E S Yeung, J. Chromatogr., A 1986, 370:83-95.
[11] H Jinlan, L Huiping, L Xiaoge,《Talanta》1998,46:1-7.