http://www.chemistrymag.org/cji/2007/097032pe.htm |
Jul.10,
2007 Vol.9 No.7 P.32 Copyright |
Zhong Yun1, Xu Zhiguang2,
Zhang Weiguang2, Yin Xia2, Tan Minyu3
(
Received on Apr.16, 2007.
Abstract: Theoretical studies on zinc complexes with dithiocarbamate Zn(S2CNBz2)2 1,Zn(S2CNBz2)2Py 2 (Bz = benzyl, Py = pyridine) have been performed by density function theory at B3LYP/6-31G* level, and their IR spectra have been calculated and analyzed exactly. According to the change of energies of frontier molecular orbitals, charge distributing, components of HOMO orbital , IR spectra and other related data, it was found that by adding nitric heterocycle it would reduce the stability and symmetry of these complexes, especially weakened S-Zn bond, which had been interpreted by charge transfer, change of orbital energies and bond valence sum analysis.Keywords density function theory, dithiocarbamate, nitric heretocycle, IR spectra
Dithiocarbamate is a kind of common ligand
containing sulfur, which could form complexes with all kinds of transition metals, B,B
metals and rare earth. For the wide application in extracting metal ions[1],
lubricant industry[2] and rubber vulcanization accelerant[3], these
complexes have been more and more studied. What’s
more, these compounds could be the precursors for / materials[4]and effective
antidotes for cadmium poisoning[5].
The structures of these compounds are various. As far as
dithiocarbamate is concerned, it is easy to form dimmer, polymer with B,B metals, whose
coordination number is 4 or 5[6,7]. However, the metal ions in those compounds
have not been coordinative saturation, they could form adducts with ligands containing N,
P, and the coordination number became 5 or 6[8]. When nitric heterocycle were
introduced, the coordination number , the difference among S-M bonds length and average
S-M bond length were increasing; the symmetry and stability of complexes were decreasing;
the differences of the chemical environment among sulfur atoms in ligands have been
obvious; IR spectra became more complicated. In recent years, the researchers have more
and more interests in studying the vibration of C-S, N-C and CSS frame.
There are many studies reported on dithiocarbamate complexes in
experimental, but seldom in theory. In recent work, we have studied Zn2(S2CNBun2)2
by quantum chemistry method[9]. In this paper, zinc complex with
dibenzyldithiocarbamate Zn(S2CNBz2)2 1 and adduct
with pyridine Zn(S2CNBz2)2Py 2 (Bz = benzyl, Py =
pyridine) have been performed by density functional theory at B3LYP/6-31G* level. Through
contrasting between the energies of frontier molecular orbital, charge distribution and
components of HOMO orbital, the authors found that after introduced pyridine, the
molecular symmetry had been destroyed, and the Zn-S bond length became longer for the
weakening of the ZnS2CN conjugation. The pyridine in the molecular form had
tendency to loose. The calculated IR spectra of complexes , which was similar to actual,
have been interpreted.
1 EXPERIMENTAL AND CALCULATE METHOD
1.1 Experimental
The complexes have been synthesized according to reference 10.
The X-ray crystal data was collected on an CCD area detector with
graphite-monochromated Mo Kα radiation (l= 0.071073nm) at 295(2)K, using the
1.2 Calculating method
Molecular structures are derived from single crystal X-ray diffraction[10]. The vibrational frequency have been calculated by B3LYP/6-31G* level of density function theory. All the calculations were performed using the Gaussian03[11] programs package.
2 RESULT AND DISCUSSION
Complex 1 is orthorhombic with space group Pbcn. Complex 2 is
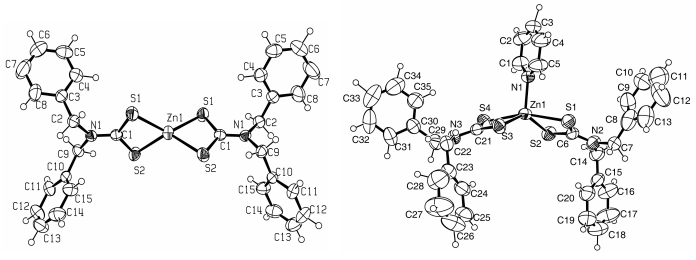
triclinic with space group P-1. Fig 1 shows the molecular structure of complexes, and
table1 shows the selected bond length and bond angle of complexes.
Complexes 1([Zn(S2CNBz2)2])
Complexes 2([Zn(S2CNBz2)2Py])
Fig.1 The structures of complexes 1 and 2
Table 1 The selected bond length(nm) and bond angle(°)
Zn(S2CNBz2)2 |
Zn1-S1 0.23343(8) |
Zn1-S2 0.23636(8) |
C1-S1 0.1730(3) |
C1-S2 0.1726(3) |
||
C1-N1 0.1329(3) |
C9-N1 0.1473(3) |
C2-N1 0.1479(3) |
||||
S1-C1-S2 117.45(15) |
S1-C1-N1 121.30(19) |
N1-C1-S2 121.26(19) |
||||
C1-N1-C9 122.4(2) |
C1-N1-C2 122.8(2) |
C2-N1-C9 114.9(2) |
||||
Zn(S2CNBz2)2Py |
Zn1-N1 0.2044(4) |
Zn1-S1 0.26183(15) |
Zn1-S2 0.23662(15) |
|||
Zn1-S3 0.23644(14) |
Zn1-S1 0.25371(14) |
C6-S1 0.1713(5) |
||||
C6-S2 0.1705(5) |
C21-S3 0.1716(5) |
C21-S4 0.1712(5) |
||||
C6-N2 0.1340(6) |
C14-N2 0.1449(8) |
C7-N2 0.1471(8) |
||||
C21-N3 0.1334(6) |
C29-N3 0.1473(6) |
C22-N3 0.1475(6) |
||||
S1-C6-S2 118.8(3) |
N2-C6-S2 121.2(4) |
S1-C6-N2 120.0(4) |
||||
S3-C21-S4 118.2(3) |
N3-C21-S4 121.4(3) |
S3-C21-N3 120.3(4) |
||||
C7-N2-C14 115.7(5) |
C7-N2-C6 121.5(5) |
C6-N2-C14 123.0(5) |
||||
C29-N3-C22 114.3(3) |
C21-N3-C22 123.8(4) |
C29-N3-C21 121.7(4) |
||||
2.1 The stability of complexes
Energies of frontier molecular orbital in complexes 1 and 2 were illustrated
in Table 2. The energies of occupational molecular orbital of complexes are all negative,
and HOMO energies of complexes are lower, -0.215a.u.(complex 1) and
-0.177a.u.(complex 2). It demonstrated that the complexes 1 and 2 are
too stable to lose electron. The
Table 2 Energies of frontier molecular orbital in complexes 1 and 2 (a.u.)
Complexes 1 |
Complexes 2 |
|||
151a |
-0.250 |
172a |
-0.236 |
|
152a |
-0.250 |
173a |
-0.229 |
|
153a |
-0.245 |
174a |
-0.225 |
|
154a |
-0.245 |
175a |
-0.222 |
|
155a |
-0.227 |
176a |
-0.210 |
|
156a |
-0.223 |
177a |
-0.205 |
|
157a |
-0.222 |
178a |
-0.197 |
|
158a(HOMO) |
-0.215 |
179a(HOMO) |
-0.177 |
|
159a(LOMO) |
-0.029 |
180a(LOMO) |
-0.054 |
|
| DEHOMO-LUMO | 0.186 |
DEHOMO-LUMO | 0.123 |
|
2.2 Natural charges distribution of atoms in
the complexes
Table 3 shows the natural charges distribution of atoms in the complexes. It is indicated
from Table 3:
1. Obvious covalent character: The valence of Zn atoms in two complexes
are both +2, but the natural charges of Zn atoms, all less than +1, are 0.481 and 0.564,
respectively. It demonstrated that obvious covalent bond have been formed between Zn atom
and ligand.
2. Natural charges distribution of S2CNC2 motion in
dithiocarbamate exhibits polarity alternation: Under the effect of polar atoms S, N, the
natural charges distribution of S2CNC2 motion in dithiocarbamate
exhibits polarity alternation along (S,S)-C-N-(C,C). And, in complex 2, the C atoms
in pyridine also exhibit polarity alternation along N1-C1-C2-C3-C4-C5.
3. Natural charges distribution incline to equilibration: The absolute
values of all atoms' natural charges are below 1. It
indicated that natural charges distribution of complexes incline to equilibration because
it exhibits polarity alternation.
When pyridine was introduced, the natural charge of Zn atom has been
more positive; natural charges of ligands(DTC) have been more negative; and natural charge
of pyridine in complex 2 was positive(see Table 3). It demonstrated that electrons
had transferred from Zn to DTC; the covalent character of Zn-S has been decreased, and
weakened; Zn-S bond length has been extended, meanwhile C-S has shortened and
strengthened. That the natural charge of pyridine in complex 2 is positive
indicates that combination between pyridine and Zn is not so stable, and the pyridine in
complex 2 would lose under the circumstance[11]. In addition, the
introduced pyridine made the natural charges of S atoms in DTC different, which increased
the asymmetry of DTC.
Table3 Natural charges of some atoms or groups in complexes
Complex 1 |
Zn1: 0.481, S1: -0.176, S2: -0.172, C1: -0.041, |
Complex 2 |
Zn1: 0.564, S1: -0.274, S2: -0.186, C6: -0.025, S3: -0.186, S4: -0.258, C21: -0.028,
N3:-0.350, N1: -0.506, C1: 0.133, C2: -0.164, C3:
-0.089, |
2.3 The bond character in complexes
The square sums of all kinds of atom orbital coefficients have been added, and been
normalized. The results are the contribution of atom orbital in HOMO orbital or components
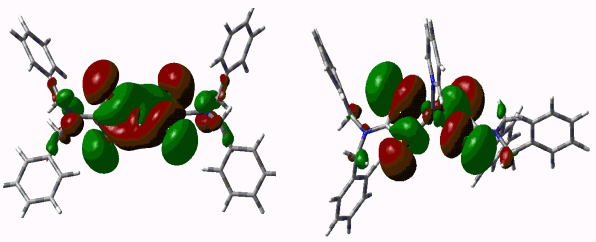
of HOMO orbital(see Table 4). Fig 2 shows HOMO orbital of complexes 1 and 2.
HOMO orbital of complexes are π bond
which was composed of multitudinous p(p) orbital of S and little p(p) orbital of N, C(see Table 4). HOMO orbital of complex 2
is a little distorted d-p antibonding orbital, and its energy is -0.17701a.u.. HOMO orbital of complex 1 is torsional d-p
antibonding orbital. Its energy(-0.21516 a.u.) is lower than complex 2 because of
partly overlapping between S atoms and Zn atom(see Fig 2).
The s orbital population of HOMO orbital in complex 2 is
higher than complex 1, which imaged that the symmetry of complex 2 was lower
than complex 1. The p(p)
orbital of N, C of HOMO in complex 2 is lower than complex 1, and p(p) orbital of S of HOMO in complex 2
is higher than complex 1. It indicated that the introduced pyridine had destroyed
conjugate of ZnS2CN, made electrons congregate on S atoms, which agreed with
natural charges distribution. From crystal x-ray analysis, Zn-S,C-N bond length in complex
2 is longer than complex 1, and S-C is shortened and strengthened.
Zn(S2CNBz2)2
Zn(S2CNBz2)2Py
Fig. 2 HOMO orbital of the complexes
Table4 The components of HOMO orbital in complexes calculated by B3LYP
Atom orbital |
Zn |
S |
N |
C |
H |
|||||
s |
p |
d |
s |
p |
s |
p |
s |
p |
s |
|
Complex 1 |
<0.01 |
2.33 |
4.76 |
0.02 |
79.17 |
<0.01 |
6.99 |
1.18 |
4.96 |
0.47 |
Complex 2 |
0.09 |
0.16 |
4.16 |
0.79 |
88.01 |
0.36 |
0.50 |
1.12 |
3.39 |
1.04 |
2.4 The effect of S-Zn by introducing nitric
heterocycle
Bond Valence Sum Analysis (BVS) is applied to complexes of zinc and cadmium dithiocarb-
amates to estimate the effective valences of the metal ions from the bond lengths reported
from their crystal structures. The valence vij of a bond between two atoms i and j is
defined so that the sum of all the valences from a given atom i with valence Vi obeys Vi =
Vij, vij = exp[(Rij-dij)/B]. Here,
The steric effect of ligands containing N have more effect to S-Zn in complexes. Since N-Zn bond length is shorter than S-Zn bond length, the ligands containing N are more approach than dithiocarbamate. For the steric effect of ligands containing N, the conjugate of MS2CN would be destroyed, and the coordinative circumstance of S atoms in ligands would be difference. It will extend S-Zn, and destroy primary dimmer, polymer structure(see Table 5). In xanthate complexes which is similar to dithiocarbamate complexes, this situations will be occurred. Adding pyridine and 1,10- phenanthroline(phen) to polymer [Cd(S2COC4H9-n)2]n(average S-Cd:0.2551nm) will acquire two monomers, [(Py)Cd(S2COC4H9-n)2][13] (average S-Cd:0.2687nm) and [(phen)Cd(S2COC4H9-n)2][14] (average S-Cd:0.2676nm), respectively.
Table 5 Average S-Zn bond length in Zinc complexes with dithiocarbamate and their nitric heterocycle adducts (nm)
Complexes |
Coordination number |
Average S-Zn bond length |
Reference |
[Zn(S2CNEt2)2]2 |
4 |
0.2383 |
15 |
[Zn(S2CNBun2)2]2 |
4 |
0.2361 |
16 |
[Zn(S2CNBz2)2] |
4 |
0.2349 |
10 |
a- [{O(CH2)4NH}Zn(S2CNEt2)2] |
5 |
0.2471 |
17 |
b- [{O(CH2)4NH}Zn(S2CNEt2)2] |
5 |
0.2468 |
17 |
[(phen)Zn(S2CNBun2)2]2 |
6 |
0.2523 |
18 |
[PyZn(S2CNBz2)2] |
5 |
0.2472 |
10 |
[(4,4'-Py)Zn(S2CNBz2)2] |
5 |
0.2464 |
19 |
Table 6 Mainly calculated and experimental IR vibration peak value of complex 1(cm-1)
Calculated |
Experimental |
|
C-N |
1549.1(1489)vw |
1489s |
|
1224.49(1177)m |
1157m |
C-S |
1027.5(987.7)m |
987.8m |
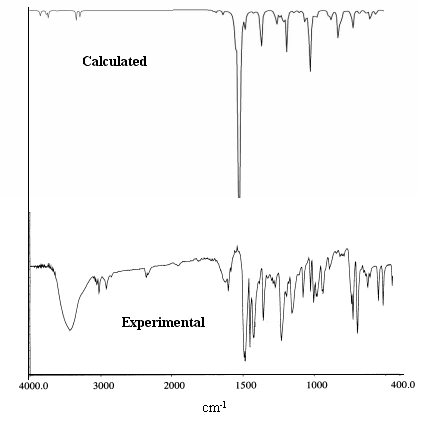
Fig. 3 The calculated and experimental IR spectra of complex 1
(The calculated frequency will be multiplied by correction factor: 0.9613)
2.5 IR spectra
Fig 3 illustrates the calculated and experimental IR spectra of complex 1. Table 6
shows the mainly calculated and experimental IR vibration peak value of complex 1.
Fig 4 illustrates the calculated and experimental IR spectra of complex 2. Table 7
shows the mainly calculated and experimental IR vibration peak value of complex 2.
From crystal x-ray analysis, it is known that complex 1 has
higher symmetry than complex 2. Two ligands in complex have little difference. The
C-N vibration appears in 1549.1 (1489)cm-1 and 1544.64 (1485) cm-1(value
in parentheses have been multiplied by correction factor: 0.9613[20]), and the
experimental value is 1489 cm-1 and 1485 cm-1. It demonstrates that
C-N have double bond character, which is in accordance with existing reports. The
vibration of ![]() appears in 1224.49 (1177)cm-1
and 1199 (1152) cm-1, and the experimental value is 1157 cm-1, which
reveals obvious single bond character. It indicates that the electrons have only
delocalized in S2CN. The C-S vibration has little split, appears in 1027.5
(987.7)cm-1 and 1026.55 (986.8)cm-1, and experimental value is only
one: 987.8cm-1. It indicates that the chemical circumstances of S atoms in
ligands have little difference.
appears in 1224.49 (1177)cm-1
and 1199 (1152) cm-1, and the experimental value is 1157 cm-1, which
reveals obvious single bond character. It indicates that the electrons have only
delocalized in S2CN. The C-S vibration has little split, appears in 1027.5
(987.7)cm-1 and 1026.55 (986.8)cm-1, and experimental value is only
one: 987.8cm-1. It indicates that the chemical circumstances of S atoms in
ligands have little difference.
The symmetry of complex 2 is
lower. Two dithiocarbamate ligands have greater difference, and the IR spectra is more
complicated. The C-N vibration appears three peaks, 1554.54 (1494) cm-1,
1531.68 (1472) cm-1, 1498.57 (1441) cm-1, and experimental value is
1494 cm-1, 1470 cm-1, 1450cm-1. In two S2CNC2
frame of complex 2, the asymmetry of A ( C6-S1: 0.1713nm, C6-S2: 0.1705nm,
C14-N2:0.1449nm, C7-N2:0.1471nm ) is higher than B ( C21-S3: 0.1716nm, C21-S4:0.1712nm,
C22-N3:0.1475nm, C29-N3:0.1473nm ) and complex 1( C1-S1: 0.1730nm, C1-S2:0.1726nm,
C9-N1:0.1473m、C2-N10.1479 ). And the polarization of
A( S1: -0.274, S2: -0.186, C6: -0.025, N2:-0.372, C7: -0.252, C14: -0.170) is also higher
than B ( S3: -0.186, S4: -0.258, C21: -0.028, N3:-0.350, C22: -0.212, C29: -0.239) and
complex 1 (S1: -0.176, S2: -0.172, C1: -0.041, N1:-0.363, C2: -0.230, C9: -0.183).
These higher asymmetry and polarization induce split in C-N vibration of A. The ![]() vibration also splits, which appears in 1330.39
(1279)cm-1 and 1221.49 (1174) cm-1, and experimental value is 1274cm-1
and 1158cm-1. It indicates that the electrons have greater delocalized area in
A, which induces C14-N2 (0.1449nm) was closer than other three C-N (0.1471nm, 0.1475nm,
0.1473nm ) and splits the
vibration also splits, which appears in 1330.39
(1279)cm-1 and 1221.49 (1174) cm-1, and experimental value is 1274cm-1
and 1158cm-1. It indicates that the electrons have greater delocalized area in
A, which induces C14-N2 (0.1449nm) was closer than other three C-N (0.1471nm, 0.1475nm,
0.1473nm ) and splits the ![]() vibration.The C-S
vibration has also three peaks, 1058.79 (1018) cm-1, 1040.01 (999.8)cm-1,
1025.42 (985.7)cm-1,and the experimental
value is 1028 cm-1, 998cm-1, 985cm-1. It demonstrates
that the chemical circumstances of S atoms are not the same. From crystal x-ray analysis
data, the C-S bond length could be considered as three groups, 0.1716nm, 0.1705nm,
0.1712nm and 0.1713nm; from natural charges distribution, they could also be considered as
three groups, -0.274,-0.258,-0.186.
vibration.The C-S
vibration has also three peaks, 1058.79 (1018) cm-1, 1040.01 (999.8)cm-1,
1025.42 (985.7)cm-1,and the experimental
value is 1028 cm-1, 998cm-1, 985cm-1. It demonstrates
that the chemical circumstances of S atoms are not the same. From crystal x-ray analysis
data, the C-S bond length could be considered as three groups, 0.1716nm, 0.1705nm,
0.1712nm and 0.1713nm; from natural charges distribution, they could also be considered as
three groups, -0.274,-0.258,-0.186.
Table 7 Mainly calculated and experimental IR vibration peak value of complex 2(cm-1)
| Calculated | Experimental |
|
C-N |
1554.54(1494)s |
1494s |
|
1330.39(1279)m |
1274w |
C-S |
1058.79(1018)m |
1028m |
valuse in parentheses have been multiplied
by correction factor: 0.9613
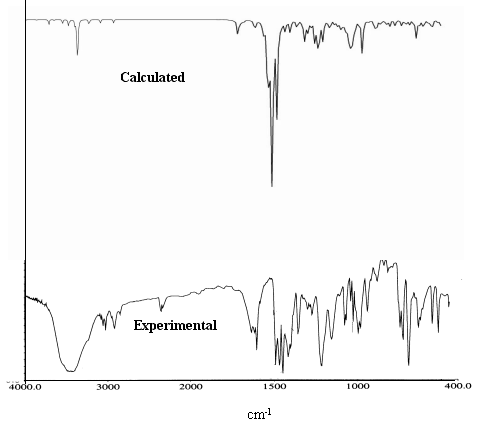
Fig 4 The calculated and experimental IR spectra of complex 2
(The calculated frequency will be multiplied by correction factor: 0.9613)
3 CONCLUSION
Introduced nitric hetercycle have increased
the coordinate number of zinc complexes with dithiocarbamate, destroyed the conjugate of
ZnS2CN, weakened the Zn-S, extended Zn-S, C-N and strengthened C-S. The
complexes are more stable because the occupational molecular orbital of complexes are all
negative and lower. It indicates that zinc complexes with dithiocarbamate could react with
nitric heterocycle to form adducts. In complex 2, the introduced pyridine has
destroyed the symmetry of complex, increased the asymmetry and polarization of S2CNC2
in ligands, induced greater difference among S atoms. The IR spectra of complex 2
is more complicated, and the vibrations of C-N,C-S and ![]() have all
split.
have all
split.
REFERENCES
[1] Wai C M, Wang S F , Liu Y.
Talanta ,1996, 432, 2083-2091
[2] Ren T H, Xia J, Zhong Y et al.
Tribology (Mocaxue Xuebao),1998, 18(3),
268-271
[3] Jiang T, Zhang W G, Shen J Y. Chinese Rare Earths (Xitu), 2000, 21(3), 39-41
[4] Zeng D, Hampden-Smith M J, Alam T M. Polyhedren, 1994, 13, 2715-2730
[5] Gale G, Atkins L M, Smith A B et al. Ann. Clin. Lab. Sci., 1984, 14, 137-145
[6] Yin X, Zhang W G, Zhang Q J, et al, Bis[bis(N,N-dibenzyldithiocarbamato)cadmium()],
Appl. Organmetallic. Chem., 2004, 18, 139-140
[7] Zhong Y, Zhang W G, Fan J et al. Acta Cryst. E., 2004, 60, 1633-1635
[8] Thirumatan S, Ramalingam K, Bocelli G et al. Polyhedron, 1999, 18, 925-930
[9] Xu X, Xu Z G, Chen Z X et al. Chin. J. Inorg. Chem (Wuji Huaxue Xuebao)., 2005,
21(7), 1049-1054
[10] Zhong Y, Zhang W G, Zhang Q J et al. Acta Chim. Sinica, (Huaxue Xuebao), 2003,
61(11), 1823-1838
[11] Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel et al.
Gaussian, Inc., Pittsburgh PA, 2003.
[12] Manohar A, Ramalingam K, Bocelli G et al. Polish J. Chem., 2001, 75, 147-151
[13] Jiang X H, Zhang W G, Zhong Y et
al. Chin. J. Inorg. Chem. (Wuji Huaxue Xuebao),2002, 18, 615-618
[14] Zhang W G, Jiang X H, Zhong Y et al. Coll.Czech.
chem..com., 2002, 67, 1623-1628
[15] Bonamico M, Mazzone G, Vaciago A et al. Acta Cryst., 1965, 19, 898-909
[16] Zhang W G, Zhong Y, Tan M Y et al. Molecule, 2003,8,411-417
[17] Ivanov A V, Kritikos M, Antzutkin O N et al. Inorg. Chim. Acta, 2001,321,
63-74
[18] Bell N A, Johnson E, March L A et al. Inorg. Chim. Acta, 1989,156, 205-211
[19] Yin X, Zhang W G, Fan J et al. Appl. Organmetallic. Chem., 2003,17, 889-990
[20] Foresman J B, Frisch E. Exploring Chemistry with Electronic Structure Methods, 2nd
Edition, Pittsburgh (USA),1996, 64.
含氮杂环对二硫代氨基甲酸类金属配合物晶体结构的影响
钟昀1 徐志广2 章伟光2
殷霞2 谭民裕3
(1 华东交通大学 基础科学学院南昌 330013; 2华南师范大学 化学与环境学院
广州 510631;3
兰州大学 化学化工学院 兰州 730000)
摘要 用密度泛函方法(DFT)对二硫代氨基甲酸锌配合物Zn(S2CNBz2)2 1,Zn(S2CNBz2)2Py 2 (Bz=苄基,Py=吡啶) 进行了研究,准确计算并分析两者的红外光谱,通过前线轨道能量、电荷布局、HOMO轨道成分分析和红外光谱对比,结合同类报道,发现含氮杂环配体的引入使得配合物的稳定性、对称性都有所下降,尤其是S-Zn键被明显削弱。以电荷转移、轨道能量变化及价键分析合理地解释该现象。
关键词 密度泛函,
二硫代氨基甲酸,含氮杂环,红外光谱