http://www.chemistrymag.org/cji/2007/098037ne.htm |
Aug.28,
2007 Vol.9 No.8 P.37 Copyright |
Analysis of monosaccharide composition in coking malt by precolumn derivatization-high performance liquid chromatography
Zhou Jianke,Han Kang,Zhang Li,Shi Zhihong
(College of Chemistry and Environmental Science, Hebei University; Key Laboratory of
Analytical Science and Technology of Hebei Province, Baoding, 071002, China)
Received on Jul.12, 2007.
Abstract In this paper, a method for the determination of monosaccharide composition in
coking malt by HPLC was established. The derivatization of monosaccharide with
1-phenyl-3-methyl-5-pyrazolone (PMP) was simplified. Four monosaccharide derivatives have
been well separated by using C18 column. The mobile phase consisted of
acetonitrile-ammonium acetate buffer (pH5.5) (71.5:28.5, v/v) and the detection wavelength
was set at 250nm. The linear ranges of mannose, rhamnose, glucose and xylose were 0.10
mmol/L-0.001 mmol/L and correlation coefficients ranged from 0.9995-0.9999. The RSD values
were all below 3.5% and the average recoveries ranged from 84.2%-108.8%.
Keyword Precolumn
derivatization; High performance liquid chromatography; Coking malt; Monosaccharide
1. INTRODUCTION
As early as in 1994, acrylamide was listed as a "potential human carcinogen"
by the International Agency for Research on Cancer (IARC). From then on, acrylamide
attracted the extensive concerns from the scientists all over the world. So far, the
formation mechanism of acrylamide in food is not very clear. But it is generally believed
that acrylamide is produced primarily at high-temperature through the Maillard reaction in
food which is rich in starch [1]. Coking malt, a kind of commonly used Chinese
herb, is rich in starch and is processed from malt at high temperature. Trace acrylamide
has been detected in it by our research group. The relationship between acrylamide and
monosaccharide composition need to be further studied. This paper described the
determination of the monosaccharide composition in processed Chinese herb for the first
time.
A considerable amount of researches have already been carried out in recent
years for the determination of . monosaccharides. Since monosaccharides encompass a number
of homologues having very similar structures and many of them exist concurrently in the
samples, monosaccharide analysis inevitably requires high-resolution separation
techniques. However, these compounds generally have low intrinsic UV spectral activity.
Therefore the derivatization of monosaccharide is
indispensable to obtain highly sensitive detection [2]. The
reagent 1-phenyl-3-methyl-5-pyrazolone (PMP) is one of the popular derivatising
agent that can react with reducing monosaccharide under
mild conditions, requiring no acid catalyst and causing no desialylation and
isomerization. PMP yields strong UV absorbance at 245 nm. This derivatization method was
first developed for the analysis of monosaccharide by Honda [3]. PMP
derivatization increases the hydrophobicity of monosaccharide, therefore HPLC is quite
suitable for analyzing PMP derivatives [4]. In this paper, on the basis of
previous research work, the RP-HPLC analysis method is established to detect
monosaccharide composition in coking malt by simplified sample processing and
derivative method.
2. EXPERIMENTAL
2.1. Apparatus and reagents
The HPLC system consisted of LC-10AT solvent delivery pump, 7725i injector and SPD-10A
UV-VIS detector from Shimadzu Corporation (Japan).The N-2000 Double-Channel
Chromatograph Data Operation Station and Software were purchased from Intelligent
Information Engineering Research Institute of Zhejiang University. The UV-265
spectrophotometer was from Shimadzu Corporation (Japan). XW-80A Vortex was from
Instrumental Factory of Shanghai Medical University.
PMP was purchased from Beijing Purchasing and Supplying Station of Chinese
Pharmaceutical Companies (China). Trifluoroacetic acid (TFA) was obtained from Kermel
(Tianjin, China). HPLC grade solvents such as methanol and acetonitrile were used to
prepare the mobile phase. Trichloromethane, Ammonia water,
ammonium acetate were analytical grade reagent. D-xylose (Xyl) and L-rhamnose (Rham) were
purchased from Beijing Chemical Reagent Company (Beijing, China), D-glucose (Glu) from
Kermel (Tianjin, China), D-mannose (Man) from Shanghai Chemical Reagent Company (Shanghai,
China). These monosaccharide reagents were all of analytical
grade.
2.2 Chromatographic conditions
Separation was performed using Diamonsil C18 (250*4.6mm i.d., 5mm) column. The
temperature of the column was kept at room temperature. The optimized mobile phase was
acetonitrile-100mmol/L ammonium acetate buffer (pH 5.5) (21.5:78.5, v/v). The detection
was carried out at the wavelength of 250 nm. The flow rate was 1.0ml/min and the injection
volume was 5.0ml.
2.3 Hydrolysis of polysaccharide
Coking malt was grinded and 2.0 g of the sample was
accurately weighed and placed in a small beaker, then 20mL of twice-distilled water was
added. After boiling for 5 min, the solution was ultrasonic extracted for 10 min, and then
stood for 10 min. Supernatant was filtered through filter paper. The leftover was washed
for three times. Then supernatant was merged and diluted to 20 ml with water. The solution
was finally filtered through 0.45mm membrane. 100 mL sample solution was mixed with 4mol/L trifluoroacetic
acid (100mL)
in frosted cone pipe with stopper (2ml). It was sealed under a nitrogen atmosphere and
kept at 110 ºC for 2h in a temperature controlled aluminum heater .
2.4 Preparation of PMP derivatives
2.4.1 Derivatives of standard monosaccharide mixture
Man (0.0180g), Glu (0.0180g), Rham (0.0182g) and Xyl (0.0150g) were all dissolved in
ammonia water (5ml) [5]. A 100mL of this solution was mixed
with a 0.5mol/L methanol solution of PMP (100μl), and the mixture was allowed to react at 70 ºCfor 30 min.
After cooling to room temperature, the reaction mixture was dried by a nitrogen stream at
100 ºC for
discarding ammonia. The residue was dissolved in distilled water and trichloromethane (1ml
each). The organic phase was discarded after vigorous shake and centrifugation (3000
r/min) to remove the excess reagents. This extraction process was repeated three times,
and then the aqueous layer was diluted with 20 times water before HPLC analysis.
2.4.2 Derivatives of
hydrolysate of polysaccharide and analysis
The product of hydrolytic reaction mentioned in 2.3 was dried by a nitrogen stream at 100
ºC. The residue was then dissolved with 100μL
ammonia liquor and 0.5mol/L methanol solution of PMP (100ml). Then the mixture was treated using the method mentioned in
2.4.1. Finally, aqueous layer was diluted 20 times for HPLC analysis.
3. RESULTS AND DISCUSSION
3.1 Hydrolysis and derivatization of polysaccharide
Hydrolysis reaction was often performed in an ampoule in other references [4].
In our method, the frosted cone tube was sealed with sealing membrane under a nitrogen
atmosphere, so the troublesome step of sealing with alcohol spray torch was omitted.
Meanwhile, experimental cost was lowered. In the literature [2], aqueous HCl
was needed to neutralize aqueous NaOH after derivative reactions. While in our experiment,
using ammonia liquor instead, the excess ammonia was simply removed under nitrogen stream
which avoided the neutralization procedure and the drying step under vacuum [5].
3.2 Selection of mobile phase
Derivatives of monosaccharides could not be well separated using the mixture of
acetonitrile-water as mobile phase. The optimized mobile phase was acetonitrile-100mmol/L
ammonium acetate buffer. The volume of acetonitrile influenced the separation greatly when
the concentration of acetonitrile increased from 20% to 22%. Considering both the
separation and retention time, the volume of acetonitrile was chosen to be 21.5% for
further study.
3.3 Linear equations, correlation coefficients and detection limits
A series of working solutions were prepared by diluting the mixed standard solution
containing four kinds of monosaccharide (0.02mmol/mL each). They were treated using the
method mentioned in 2.4.1 and the final solution was detected under the optimized
chromatographic conditions. The calibration graphs of the standard compounds were found to
be linear over the concentration range studied. The linear equations and the detection
limits of the derivatives are listed in Table 1.
Table 1 Linearity and the detection limits of PMP derivatives of four kinds of monosaccharide
Monosaccharide |
Linear equation |
Correlation coefficient(n=5) |
Detection limit |
Mannose |
Y=3.26×106X+2965.42 |
0.9999 |
0.001 |
Rhamnose |
Y=3.36×106X+13241.83 |
0.9999 |
0.002 |
Glucose |
Y=2.57×106X+4469.27 |
0.9995 |
0.002 |
Xylose |
Y=3.36×106X-55.20 |
0.9999 |
0.002 |
Y-peak area; X-the concentration of the detected compound
3.4 Recoveries and precision
The recoveries of the method were studied. The standard solutions of different
concentrations were added into coking malt sample solutions. Then the spiked sample
solutions were treated with the same method as described in 2.3 and 2.4. The
concentrations of Man, Rham and Xyl were examined under the optimized chromatographic
conditions and the recoveries could be calculated according to the linear equation. The
precision of the method was determined by preparing the sample with the same method as
described in 2.3 and 2.4 for each analysis, and RSD values were determined by 5 repeated
analyses. As for Glu, the coking malt sample solution was diluted 100 times to carry on
the similar operation again. The results are given in Table 2.
Table 2 Recoveries and precision
Monosaccharide |
Sample contents |
Added contents |
Determined contents |
Recoveries |
RSD /% (n=5) |
Mannose |
1.28 |
4.50 |
6.17 |
108.71 |
1.16 |
Rhamnose |
0.71 |
4.55 |
4.92 |
92.51 |
1.09 |
Glucose |
147.15 |
450.4 |
526.14 |
84.15 |
2.03 |
Xylose |
1.56 |
3.75 |
5.44 |
103.41 |
3.28 |
3.5 Analysis of coking malt samples
Coking malt samples were analyzed using the established method, and the chromatograms are shown in Figure 1.The use of merely monosaccharide standard compounds which the laboratory have had, four monosaccharide were identified by the retained value qualitative method, they were Man, Rha, Glu and Xyl. The molar ratio of Man, Rha, Glu and Xyl in coking malt was 1.82:1.00:209.44:2.67 by HPLC analysis and molar ratio calculation. The RSD values of four kinds of monosaccharide were all lower than 3.5%. The repeatability of detected results was satisfactory.
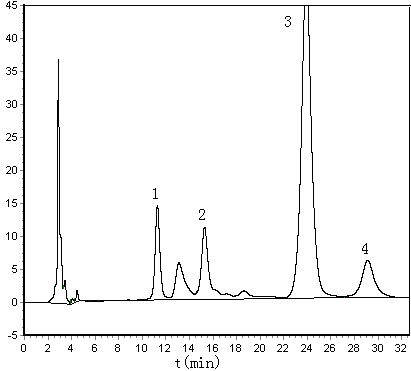
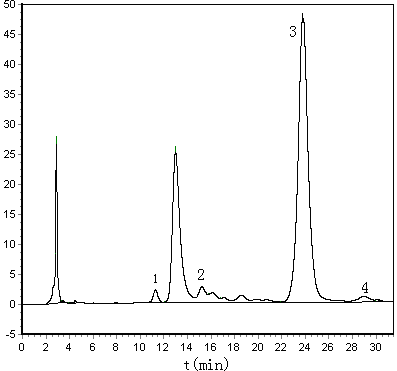
Figure 1 Chromatograms of spiked
coking malt sample (A) and coking malt sample (B)
1-Mannose; 2-Rhamnose; 3-Glucose; 4-Xylose
4. CONCLUSIONS
A simple, rapid and reproducible method for the derivatization of monosaccharide by
PMP was established, which avoided the procedures of the drying of hydrolysates and
derivative solution. The experimental results demonstrated that the developed method was
suitable for the analysis of monosaccharides by HPLC.
ACKNOWLEDGEMENT This work was supported by the National Natural Science Foundation of China (No. 20575016).
REFERENCES
[1] Zhang Y L, Xia W S. Food Research and Development (Shipin Yanjiu Yu Kaifa), 2006,
27 (4): 134-136.
[2] Xu J, Zhang L Y, Zhang Q H et al. Chinese Journal of Chromatography (Se Pu), 2003, 21
(4): 3-366.
[3] Honda S, Akao E, Suzuki S et al. Analytical Biochemistry, 1989, 180 (2): 351-357. 36
[4] Ma D Y, Chen J, Li P et al. Chinese Journal of Analytical Chemistry (Fenxi Huaxue),
2002, 30 (6): 702.
[5] Lin X, Wang Z F, Huang L J et al. Chemical Journal of Chinese Universities (Gaodeng
Xuexiao Huaxue Xuebao), 2006, 27 (8): 1456-1458.
柱前衍生化高效液相色谱法测定焦麦芽中的单糖组成
周建科,韩 康,张 立,石志红
(河北大学化学与环境科学学院, 河北省分析科学技术重点实验室, 河北保定 071002)
摘要 用高效液相色谱法对高温炮制药材焦麦芽中的单糖组成进行测定,简化了PMP衍生步骤。在最佳色谱条件下四种单糖衍生物得到了较好的分离。色谱柱为C18色谱柱,流动相乙腈-100mmol/L醋酸铵缓冲溶液(PH5.5)(21.5:78.5),250nm检测。在0.10mmol/L-0.001mmol/L范围内,甘露糖、鼠李糖、葡萄糖和木糖的线性相关系数在0.9995-0.9999之间,相对标准偏差在3.5%以下,平均回收率在84.2%-108.8%之间。
关键词 柱前衍生;反相高效液相色谱;焦麦芽;单糖