http://www.chemistrymag.org/cji/2007/09a046nc.htm |
Oct.14,
2007 Vol.9 No.10 P.46 Copyright |
Zhu Tao1, Yang Gengliang1,2, Cao Weimin1,
Zhao Yu1, Li Fengxin1
(1College of pharmacy, Hebei University, Baoding 071002; 2Molecular
Science center, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080)
Keywords Weak ion exchange monolithic column; On-line SPE; Cefazolin sodium; Plasma 弱离子整体柱在线固相萃取及分析人血浆中头孢唑啉钠
朱涛1,杨更亮1,2,曹伟敏1,赵玉1,李风新1
(1河北大学药学院 保定 071002;2中国科学院化学研究所分子科学中心 北京 100080)
国家自然科学基金资助项目(NO.20375010,20675084),河北省科技攻关项目(NO.06276479B,07276407D),中国科学院“百人计划”资助项目.
摘要 以甲基丙烯酸缩水甘油酯(GMA)、甲基丙烯酸(MAA)为功能单体,乙二醇二甲基丙烯酸酯(EDMA)为交联剂,在不锈钢柱管中聚合反应,然后用乙二胺在线修饰为弱离子交换整体柱。将其作为在线固相萃取材料,在水下即可去除人血浆中的大分子干扰物质,同时对带负电荷的小分子药物有富集作用,本文用该离子柱在线预处理同时测定人血浆中头孢唑啉钠。分析流动相为9.4 mmol
·L-1 Na2HPO4 和5.3 mmol·L-1 柠檬酸-乙腈(体积比87:13),流速为1.0 mL·min-1, 在254 nm处进行检测。实验结果表明,该方法具有良好的线性,回收率及精密度,避免了繁琐的样品预处理过程,为血浆中痕量药物的分析提供了一种快速、经济和有效的新方法。关键词 弱离子交换整体柱;在线固相萃取;头孢唑啉钠;血浆
血浆是血液的重要组成分,为淡黄色液体,通常对血浆等生物样品预处理的方法有酸碱或有机溶剂沉淀蛋白法
[1-3]和离线固相萃取法[4-5],但这些方法具有操作起来比较繁琐,费时费力,且引入有机溶剂,对环境不友好等缺点,而在线固相萃取可以解决这些问题。该法对痕量物质测定尤为灵敏,生物样品可以直接上样,允许去除大分子的同时富集药物,然后结合高效液相色谱等分析技术,对生物样品中的待测药物进行分析测定。H.A.G.
Niederlander等[6]结合在线固相萃取和液质联用技术分析测定了血清中的氯氮平;杨更亮等[7]利用自制预处理柱在线固相萃取分析测定了人血液和尿液中的药物,表明在线固相萃取技术在生物样品预处理方面的应用价值。
头孢唑啉钠为耐酸,耐酶半合成青霉素,对革兰氏阳性菌和阴性菌有广谱杀灭作用,又对耐青霉素的金黄色葡萄球菌有效,广泛用于人和动物细菌感染疾病的预防与治疗。此药主要从肾脏排泄,临床上在应用头孢唑啉钠的同时进行血药浓度测定,根据血药浓度计算出药物在个体体内的排泄速度,然后计算出适合个体的调整用药量,再结合肾功情况调整临床上实际用药剂量,具有一定的实用价值。本文研制了一种新型用于在线固相萃取预处理的整体柱,分析测定了人血浆中的头孢唑啉钠。
1.1 试剂与仪器
主要仪器:PU-1586高效液相色谱仪(日本,JASCO公司);UV-1570紫外可变波长检测器(日本,JASCO公司);不锈钢空色谱柱管(10 mm × 4.6 mm i.d.);HW-2000色谱工作站(南京千谱软件有限公司);ODS C18柱(DiamonsilTM,250 mm × 4.6 mm i.d.,5 μm);HH-601 超级恒温水浴(江苏省金坛市荣华仪器制造有限公司)。
主要试剂:头孢唑啉钠冻干粉(中诺药业(石家庄)有限公司);Na2HPO4(北京益利精细化学品有限公司);柠檬酸(河北省保定市化学试剂厂);乙腈,四氢呋喃,环己醇,正十二醇(天津北联精细化学品开发有限公司);乙二胺(天津市天大化工实验厂);甲基丙烯酸(MAA天津市化学试剂一厂);偶氮二异丁腈(AIBN,上海试剂四赫维化工有限公司);甲基丙烯酸缩水甘油酯(GMA,苏州安利化工厂);乙二醇二甲基丙烯酸酯(EDMA,美国Acros 公司);以上液体试剂进入色谱系统前均经过0.45 μm微孔滤膜过滤。健康人血浆由河北大学医院提供。
1.2 弱离子交换整体柱制备
在干燥的不锈钢空柱管中加入一定比例的经超声及氮气除氧的GMA、MAA、EDMA、AIBN、正十二醇和环己醇的混合溶液,抽真空20 min后密封,在55 ℃恒温条件下反应24小时,用甲醇冲去致孔剂及未反应可溶性物质后,将乙二胺-四氢呋喃混合溶液(1:1,V/V)在60 ℃恒温条件下以0.1 mL·min-1 通过色谱柱,反应24小时后用甲醇冲去未反应的乙二胺,至色谱柱流出液不显碱性。反应过程见图1。
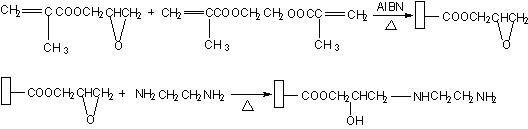
图1 弱离子交换整体柱的制备过程
Fig.1 The preparation of the ion exchange monolithic column
1.3 样品制备
准确配置1000 mg
1.4 色谱条件
弱阴离子交换整体柱(10 × 4.6 mm i.d.);分析柱为ODS C18柱;流动相为9.4 mmol·L-1 Na2HPO4 和5.3 mmol·L-1 柠檬酸 - 乙腈(体积比87:13),流速为1.0 mL·min -1, 在紫外检测波长254 nm处进行检测。
1.5 样品预处理方法与色谱分析
将预处理柱(弱阳离子交换整体柱)置六通进样阀定量环处,当处在“Load”位置时,首先推入2.0 mL过滤三蒸水,然后将50 μL待测样品直接上样,再用3.0 mL 过滤三蒸水淋洗,最后将六通进样阀扳至“Injection”位置,进入C18分析柱进行定量分析。 2 结果与讨论
2.1 实验条件的优化
2.1.1 MAA/(GMA+MAA)的单体比例对弱离子交换整体柱性能的影响
头孢唑啉钠(Pka,2.5)和血液中大分子如白蛋白(PI,4.9),与预处理柱上的羧基和氨基两种基团间均有静电引力和排斥作用,两种静电力间的竞争决定药物和大分子在预处理柱上的保留情况。MAA/(GMA+MAA)的单体比是决定离子柱性能优劣的关键因素,其它条件不变,单体比分别为0, 5%, 10%, 15%, 20%, 30%, 40%, 实验结果表明单体比为20%时,离子柱才能达到水下保留药物同时去除大分子的效果。
2.1.2 弱离子整体柱预处理能力的考察
血浆中的蛋白种类较多且含量高,为考察预处理柱能否较好的去除血浆中的内源性干扰物质,分别进50 μL空白血浆样品和头孢唑啉钠水溶液于空管和弱离子柱,水为流动相,流速为1.0 mL·min -1,检测波长分别为280 nm和254 nm。由于头孢唑啉钠可以与预处理柱上的氨基产生较强的静电吸引作用,在整体柱上有一定的保留,而蛋白等内源性物质在预处理柱上没保留,因此,在3 min时,蛋白等内源性物质能被较好的洗脱下来,而头孢唑啉钠在整体柱上有较好的保留。实验结果表明,用3.0 mL水作为洗脱淋洗液,即可完成对血样的预处理。由于血浆中的药物浓度较低,所以先通过预处理柱对药物富集后再进行分析,本文的富集进样体积为50 μL,实验结果表明,此进样体积可以满足血浆中头孢唑啉钠的测定要求。
2.1.3 样品预处理过程的分析
本实验利用液相色谱仪上六通进样阀实现柱切换过程,进行血浆样品的预处理。首先将预处理柱置六通进样阀定量环处,当处在“Load”位置时,推入2.0 mL 过滤三蒸水,目的是去除预处理柱和连接管中的甲醇等有机溶剂,防止蛋白变性;然后将50 μL待测样品直接上样,样品从预处理柱的尾端进入,用3.0 mL过滤三蒸水淋洗,洗脱下来的蛋白由柱子顶端流出,而药物则富集在预处理柱的尾端,从而实现去除蛋白等生物内源性干扰物质和富集药物的目的;当处在“Injection”位置时,分析流动相将保留在预处理柱上的药物反向洗脱后进入C18分析柱进行定量分析。由于药物被富集在预处理柱的尾端,分析流动相是反向洗脱的,此时可以认为药物能够在很短的时间内从预处理柱中洗脱下来,这样可以有效避免传统非反向洗脱而导致药物在预处理柱中洗脱缓慢而产生的色谱峰展宽、拖尾等不良现象,改善色谱峰形。
2.1.4 血液样品的色谱分离图
图2为空白血浆和1.0 mg·L-1头孢唑啉钠血样的色谱图。头孢唑啉钠的保留时间为15.9 min,由空白血样对照可知在药物出峰处无干扰,能与前面系统峰分离开,峰形良好。说明该方法分离效果好,专属性好。
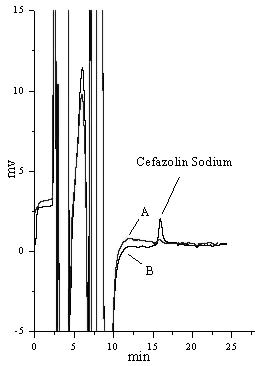
图2 空白血样(A)和头孢唑啉钠血样(B)的色谱图
2.2 方法学的考察
2.2.1 标准曲线及检出限
精密量取头孢唑啉钠标准溶液,用血浆将其依次稀释成浓度为1.0
mg
2.2.2 精密度及回收率试验
准确配制头孢唑啉钠1.0 mg
表1 头孢唑啉钠三种不同质量浓度的血液样品精密度及回收率
Table 1 The precision and recovery of cefazolin sodium determination in human plasma samples spiked with three different concentrations
Spiked concentration |
Intra-day precision RSD(%) |
Inter-day precision |
Recovery |
Absolute recovery |
1.0 |
3.2 |
3.8 |
96.7 |
83.5 |
10 |
2.1 |
2.6 |
97.3 |
80.2 |
30 |
3.8 |
3.8 |
102.4 |
86.1 |
本文研制了一种新型用于在线固相萃取预处理的整体柱,采用该离子柱在线预处理同时测定人血浆中头孢唑啉钠,方法避免了繁琐的样品预处理过程,为血浆中痕量药物的分析提供了一种快速、经济和有效的新方法。 REFERENCES
[1] Ban E, Maeng J E, Woo J S, et al. J. Chromatogr. B, 2006, 831 (1-2): 230-235
[2] Jia Y W, Wang G J, Xie H T. J. Chromatogr. B, 2007, 847 (2): 72-77
[3] Xia X R, Monteiro-Riviere N A, Riviere J E. J. Chromatogr. A, 2006, 1129 (2): 216-222
[4] Ramanathan S, Karupiah S, Nair N K et al. J. Chromatogr. B, 2005, 824 (1-2): 45-50
[5] Saracino M A, Mercolini L, Flotta G et al.. J. Chromatogr. B, 2006, 843 (2): 227-233
[6] Niederlander H A G, Koster E H M, Hilhorst M J et al. J. Chromatogr. B, 2006, 834 (1-2): 98-107
[7] Yang G L, Feng S, Liu H Y et al. J. Chromatogr. B, 2007, 854 (1-2): 85-90