http://www.chemistrymag.org/cji/2007/09c054pc.htm |
Dec.1,
2007 Vol.9 No.12 P.54 Copyright |
Yu Cheng, Zhang Xiangdong, Guan Hongyu, Ge
Chunhua, Wu Yang, Han Guangxi
(College of Chemistry, Liaoning University, Shenyang, 110036)
Abstract Support vector regression
algorithm was applied to study on quantitative structure-activity relationship of small
organic moleculars solvation free energies in aqueous solution. The eight kinds of
molecular characteristic parameters, octanol/water partition coefficient(LogP),
molecular polar surface area(TPSA), molecular volume(V), molecular weight(MW),
number of hydrogen bond acceptors(NON),number of hydrogen bond donors(NOH,NH),number of
rotatable bonds(Nrotb),number of unhydrogen
atom in a small organic molecular(natoms), were calculated. A more predictive model
was set up through optimizing kernel function parameters. The squared correlation
coefficient(r2) values between experimental versus calculated solvation free
energies for 131 molecules in the training set and 24 molecules in the test set are 0.95
and 0.94, respectively.The experimental results showed that support vector regression
algorithm would be a useful tool for rapid and accurate calculation of molecular solvation
free energies in aqueous solution.
Keywords support vector regression; solvation free energy ; quantitative
structure-activity relationship
余成, 张向东, 关宏宇, 葛春华, 吴阳, 韩光喜
(辽宁大学化学学院,辽宁 沈阳 110036)
摘要
本文计算获得了155个有机小分子化合物的正辛醇/水分配系数(LogP)、分子拓扑极表面积(TPSA)、分子体积(V)、分子质量(MW)、氢键接受体的数目(NON)、氢键给体的数目(NOH,NH)、可旋转键的数目(Nrotb)、分子所含有的非氢原子数目(natoms)8个结构特征参数。采用支持向量回归(SVR)算法,对这些有机小分子溶解自由能进行定量构效关系研究。通过核函数参数的优化,建立了预测模型,训练集和预测集的实验溶解自由能与计算溶解自由能的相关系数r2分别为0.95和0.94。研究结果表明,支持向量回归算法可用于有机小分子化合物在水溶液中的溶解自由能的快速、准确计算。关键词 支持向量回归 溶解自由能 定量构效关系
分子溶解自由能与分子在水溶液中的溶解性类似,是分子重要的的物理化学性质。在代谢反应的平衡和动力学研究、分子间的结合和结构变化等众多的化学和生物过程中,分子溶解自由能是一个非常重要的量
[1]。药物发现的早期,一个重要的任务就是计算分子溶解自由能,它影响药物在作用位点上的生物活性[2]。近年来,由于组合化学的发展,需要估算与结构相关的化合物的溶解性,这进一步促使研究更可靠、快捷的计算方法来预测有机分子的溶解自由能。特别是合成化学的快速发展,收集了大量的与药物相关的化合物,在合成药物之前需要计算它们相关的物理化学性质,如溶解自由能、亲脂性常数等,以建立药物设计数据库。然而由于实验测定溶解自由能非常费时,液液中溶质和溶剂相互作用的复杂性,溶解自由能被认为是最难以计算的能量问题。自Yalkowsky和Valvani的开创性研究以来,许多统计建模方法已用于预测分子溶解自由能,如多元线性回归[3]、人工神经网络[4]、遗传算法[5]、化学键和基团贡献等方法[6]。但以往的计算方法大多需要分子力学和分子动力学的复杂计算,或者由于计算参数过多,难以达到快速、准确计算分子溶解自由能的目的。
Vladimir N. Vapnik等通过三十余年严格的数学理论研究提出统计学习理论[7],并在统计学习理论的基础上发展了一种新的通用学习方法----支持向量机[8](Support Vector Machine或SVM),该方法包括支持向量分类(Support Vector Classification或SVC)和支持向量回归(Support Vector Regression或SVR),已在语言识别、人脸识别、文字识别、药物设计等领域得到成功应用[9-11]。本文尝试采用支持向量回归算法建立预测模型,所得预测结果优于传统统计建模方法。 1 SVR算法原理及其实现
SVM算法能较好地解决有限样本、多变量、高维数等实际问题,成功地解决了过拟合的控制问题,提高了算法的预报能力,SVR算法的具体原理可见文献[12、13]。将SVM方法应用于函数拟合问题时,即支持向量回归(SVR),SVR法的基础是引入不敏感损失函数e、惩罚参数C和核函数。不敏感损失函数e意义:当函数拟合到残差小于e时,即不再要求进一步减小残差,该算法得到的不是唯一解而是一组解,然后根据增强预报能力的要求,从中选出预报能力强的唯一解;惩罚参数C控制对超出误差e的样本的惩罚程度;核函数方法是将原始数据映射到高维空间,使非线性问题转化为线性问题求解。不敏感损失函数e、惩罚参数C和核函数可以通过优化而得到。本所有计算在奔腾2.40GHz微机上使用基于SVMDark软件改编的计算程序完成[14]。
2 计算方法
本文所选的155个有机小分子化合物的实验溶解自由能数据来自文献[5]、[6](见表1),分子结构特征参数采用Molinspiration[15]软件计算,包括8个参数:正辛醇/水分配系数(LogP)、分子拓扑极表面积(TPSA)、分子体积(V)、分子质量(MW)、氢键接受体的数目数目(NON)、氢键给体的数目(NOH,NH)、可旋转键的数目(Nrotb)、分子中所含有的非氢原子数目(natoms)。
2.2 数据的预处理
在进行SVR法计算以前将各特征参数做归一化处理,以避免训练中数值较大的特征参数所占的权重过大,缩短运算时间,同时为使计算数据表示更方便,本文采用下式对特征参数进行归一化:
式中cmax,cmin分别为某一特征参数数据中的最大值和最小值,归一化后每个特征参数计算结果保留四位有效数字。
3 结果和讨论
由于已知实验溶解自由能的有机小分子化合物的数量有限, 如何将其划分为训练集和测试集, 使所建立的模型具有较好的预测能力也是构建统计模型的关键。在建立训练样本集时,应注意各样本的多样性,这样通过训练学习,就能很好地保持了原有的支持向量机所包含的信息,而且这种学习也是可积累的,以此保证已训练好的模型对待测样本具有较强的包容能力、适应性和非常强的推广能力。本文以所计算的八个参数为输入向量,以溶解自由能为目标值。为便于与文献[5]比较,SVR法中选取的训练集和测试集与文献[5]相同。
3.2 SVR参数的影响与选择
在训练集一定的情况下,选取合适的核函数成为重点,核函数的类型决定了特征空间的结构,选择不同形式的核函数,可以构建不同的支持向量机,对应不同的算法。经格式搜索和交叉验证,选用径向基函数K(ci c)构建预测模型:
上式中g是常数,ci和c是两个独立变量,g通过调节径向基函数的形状来控制SVM 的泛化能力。SVR法中的参数主要有不敏感系数e、惩罚参数C和核函数参数g,具体选择是以回归误差最小为依据,回归误差以MSE(mean square error)表示,
MSE=
不敏感系数e反映模型对输入变量所含噪声的敏感程度,可用以控制模型的拟合精度。 e 越大,拟合精度越低,所导出的支持向量较少,模型的复杂度较低,其泛化能力较强; 反之,e越小,拟合精度越高,支持向量数增多,模型的复杂度较高,其泛化能力较低。取 C=400、g=0.5,e在0.01-0.1范围内训练,e对训练集拟合和预测集预测回归误差的影响 见图1,由图可见取e=0.05预测效果最好。
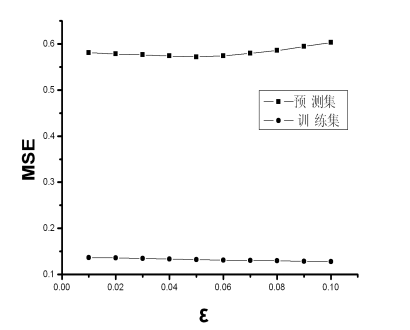
(a)
(b)
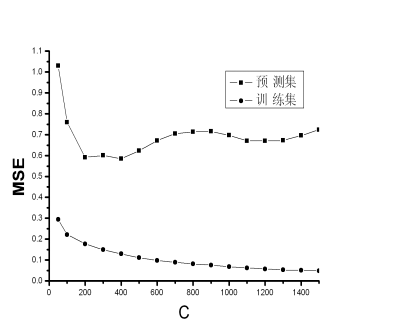
图1 e对训练集和预测集回归误差的影响(a)和C对训练集和预测集回归误差的影响(b)
Figure 1 Effect of Insensitive Factor on regression error of the training set and the test set(a),and effect of Plenalty Factor on regression error of the training set and the test set(b)
惩罚参数C是一调整参数,控制着最小经验误差和最大边界之间的均衡,C值太小时,会发生欠拟合,C值太大时,则产生过拟合。惩罚参数C的选取以回归误差最小进行选取,C对训练集拟合和预测集预测回归误差的影响见图1,由图可见C=400时,预测误差最小。
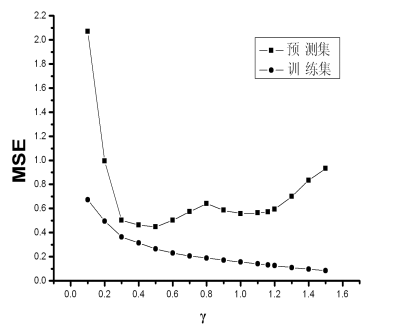
图2 g对训练集和预测集回归误差的影响
Figure 2 Effect of g on regression error of the training set and the test set
3.3 支持向量机的回归模型及预测结果
通过以上调节结果可知,表1所列的155种化合物,以LogP、TPSA、V、MW、NON、NOH,NH、Nrotb、natoms 8个变量为输入向量,选取径向基核函数,经格式搜索和交叉验证,预测模型最优参数C、e和g分别取400、0.05和0.5时,支持向量回归的模型为:
(1)式中
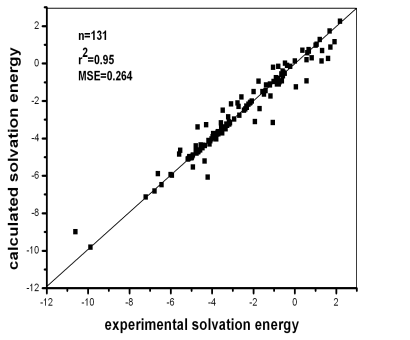
(a)
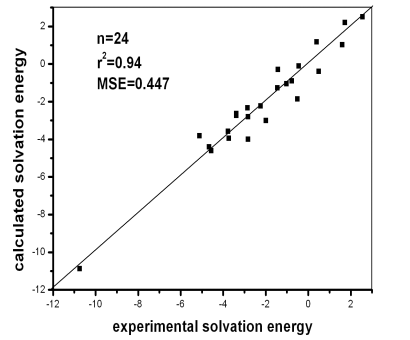
(b)
图3 训练集(a)和测试集(b)化合物的实验和计算溶解自由能的相关性
Figure 3 Correlation between
experimental versus calculated solvation free energies for (a) 131 molecules in the
training set and (b) 24 molecules in the test set. All energy values are given in
kcal/mol.
表1 训练集(a)和测试集(b)化合物的实验和计算溶解自由能( kcal/mol)
no. |
molecule name | expt |
calc |
no. |
molecule name | expt |
calc |
(a) Training Set |
|||||||
1 |
cyclopentane | 1.22 |
1.27 |
67 |
methanol | -5.14 |
-5.09 |
2 |
methylcyclopentane | 1.62 |
0.26 |
68 |
methane thiol | -1.26 |
-1.16 |
3 |
methylcyclohexane | 1.73 |
0.87 |
69 |
ethanol | -4.96 |
-4.96 |
4 |
ethylene | 1.30 |
0.12 |
70 |
2,2,2-trifluoroethanol | -4.35 |
-5.22 |
5 |
1-pentene | 1.69 |
1.73 |
71 |
1-propanol | -4.92 |
-4.89 |
6 |
cyclopentene | 0.57 |
-0.93 |
72 |
allyl alcohol | -5.10 |
-4.98 |
7 |
2-methyl-2-butene | 1.33 |
0.68 |
73 |
1,1,1-trifluoro-2-propanol | -4.21 |
-6.08 |
8 |
cyclohexene | 0.37 |
0.69 |
74 |
1-butanol | -4.78 |
-4.61 |
9 |
4-methyl-1-pentene | 1.93 |
1.15 |
75 |
2-methyl-1-propanol | -4.57 |
-4.62 |
10 |
1-methylcyclohexene | 0.68 |
0.73 |
76 |
1-pentanol | -4.55 |
-4.35 |
11 |
1-octene | 2.20 |
2.25 |
77 |
cyclohexanol | -5.02 |
-5.03 |
12 |
1,3-butadiene | 0.57 |
0.19 |
78 |
4-methyl-2-pentanol | -3.79 |
-4.03 |
13 |
2-methyl-1,3-butadiene | 0.69 |
0.64 |
79 |
phenol | -6.62 |
-5.90 |
14 |
1,5-hexadiene | 1.02 |
0.97 |
80 |
4-bromophenol | -7.20 |
-7.15 |
15 |
propyne | -0.48 |
-0.53 |
81 |
thiopheno | -2.58 |
-1.80 |
16 |
1-pentyne | 0.01 |
0.13 |
82 |
2-cresol | -5.94 |
-5.99 |
17 |
1-heptyne | 0.61 |
0.56 |
83 |
4-hydroxybenzaldehyde | -10.61 |
-8.99 |
18 |
1-nonyne | 1.06 |
1.01 |
84 |
4-tert-butylphenol | -6.00 |
-5.95 |
19 |
benzene | -0.90 |
-0.77 |
85 |
acetaldehyde | -3.55 |
-3.43 |
20 |
p-xylene | -0.82 |
-0.81 |
86 |
butanal | -3.22 |
-2.86 |
21 |
propylbenzene | -0.54 |
-0.49 |
87 |
heptanal | -2.71 |
-2.31 |
22 |
2-butylbenzene | -0.46 |
0.00 |
88 |
trans-2-butenal | -4.28 |
-3.28 |
23 |
naphthalene | -2.45 |
-2.50 |
89 |
trans-2-octenal | -3.48 |
-2.50 |
24 |
fluoromethane | -0.22 |
-0.17 |
90 |
trans-trans-2,4-hexadienal | -4.70 |
-3.40 |
25 |
trifluoromethane | 0.82 |
0.29 |
91 |
benzaldehyde | -4.08 |
-4.13 |
26 |
chloromethane | -0.54 |
-0.51 |
92 |
acetone | -3.85 |
-3.80 |
27 |
trichloromethane | -1.04 |
-0.22 |
93 |
2-pentanone | -3.56 |
-3.17 |
28 |
bromomethane | -0.80 |
-0.85 |
94 |
2-octanone | -2.92 |
-2.97 |
29 |
tribromomethane | -2.16 |
-2.11 |
95 |
acetophenone | -4.64 |
-4.67 |
30 |
iodomethane | -0.90 |
-0.85 |
96 |
acetic acid | -6.78 |
-6.83 |
31 |
chlorofluoromethane | -0.79 |
-0.16 |
97 |
butyric acid | -6.44 |
-6.49 |
32 |
chloroethane | -0.64 |
-0.56 |
98 |
methylacetate | -3.66 |
-3.71 |
33 |
bromoethane | -0.70 |
-0.93 |
99 |
ethylacetate | -3.12 |
-3.17 |
34 |
iodoethane | -0.73 |
-0.98 |
100 |
isopropylacetate | -2.68 |
-2.77 |
35 |
1,2-dichloroethane | -1.75 |
-0.96 |
101 |
isobutylacetate | -2.39 |
-2.44 |
36 |
1,2-dibromoethane | -2.13 |
-2.08 |
102 |
propylbutyrate | -2.31 |
-2.36 |
37 |
1-chloro-2-bromoethane | -1.98 |
-1.51 |
103 |
methyloctanoate | -2.07 |
-2.02 |
38 |
2-chloro-1,1,1-trifluoroethane | 0.06 |
-1.26 |
104 |
methylbenzoate | -4.34 |
-4.39 |
39 |
1-chloropropane | -0.36 |
-0.11 |
105 |
ethylamine | -4.67 |
-4.72 |
40 |
1-bromopropane | -0.57 |
-0.67 |
106 |
butylamine | -4.43 |
-4.38 |
41 |
1-iodopropane | -0.62 |
-0.94 |
107 |
hexylamine | -4.09 |
-4.14 |
42 |
cis-1,2-dichloroethylene | -1.19 |
-1.10 |
108 |
dimethylamine | -4.34 |
-4.39 |
43 |
trans-1,2-dichloroethylene | -0.77 |
-1.10 |
109 |
diethylamine | -4.12 |
-4.30 |
44 |
3-chloropropene | -0.58 |
-0.41 |
110 |
pyrrolidine | -5.54 |
-4.65 |
45 |
chlorobenzene | -1.02 |
-0.97 |
111 |
piperidine | -5.17 |
-5.12 |
46 |
bromobenzene | -1.48 |
-1.65 |
112 |
dipropylamine | -3.70 |
-3.65 |
47 |
1,2-dichlorobenzene | -1.38 |
-1.16 |
113 |
hexamethyleneimine | -4.97 |
-5.02 |
48 |
1,4-dibromobenzene | -2.33 |
-2.28 |
114 |
trimethylamine | -3.27 |
-3.32 |
49 |
p-bromotoluene | -1.41 |
-1.46 |
115 |
triethylamine | -3.07 |
-2.16 |
50 |
1-bromo-2-ethylbenzene | -1.20 |
-1.15 |
116 |
n-methylpyrrolidine | -4.02 |
-4.04 |
51 |
o-bromocumene | -0.86 |
-1.10 |
117 |
n-methylpiperidine | -3.94 |
-3.75 |
52 |
dimetyl ether | -1.92 |
-3.11 |
118 |
ethylenediamine | -9.88 |
-9.83 |
53 |
dimethyl sulfide | -1.56 |
-1.51 |
119 |
acetonitrile | -3.94 |
-3.99 |
54 |
1,3-dioxolane | -4.14 |
-4.15 |
120 |
butyronitrile | -3.69 |
-3.77 |
55 |
methylpropyl ether | -1.69 |
-2.42 |
121 |
nitroethane | -3.76 |
-3.78 |
56 |
tetrahydrofuran | -3.51 |
-3.40 |
122 |
2-nitropropane | -3.18 |
-3.13 |
57 |
dioxane | -5.11 |
-5.06 |
123 |
nitrobenzene | -4.17 |
-4.12 |
58 |
methyl tert-butyl ether | -2.24 |
-2.19 |
124 |
3-nitrotoluene | -3.50 |
-3.71 |
59 |
2-methyltetrahydrofuran | -3.34 |
-3.49 |
125 |
pyridine | -4.75 |
-4.80 |
60 |
tetrahydropyran | -3.16 |
-3.24 |
126 |
4-methylpyridine | -4.99 |
-5.04 |
61 |
dipropyl ether | -1.17 |
-1.74 |
127 |
4-ethylpyridine | -4.78 |
-4.40 |
62 |
1,2-diethoxyethane | -3.30 |
-3.25 |
128 |
2,4-dimethylpyridine | -4.92 |
-5.54 |
63 |
di-n-butyl ether | -0.84 |
-0.89 |
129 |
2-methylpyrazine | -5.58 |
-4.85 |
64 |
anisole | -1.05 |
-3.16 |
130 |
2-ethyl-3-methoxypyrazzine | -4.45 |
-4.50 |
65 |
thioanisole | -2.76 |
-2.12 |
131 |
2-isobutyl-3-methoxypyrazine | -3.73 |
-3.68 |
66 |
2,2?-dichlorodiethyl sulfide | -3.97 |
-3.92 |
||||
(b) Test Set |
|||||||
1 |
2-methylpentane | 2.56 |
2.50 |
13 |
3-hexanol | -3.73 |
-3.96 |
2 |
cis-1,2-dimethylcyclohexane | 1.60 |
1.02 |
14 |
4-nitrophenol | -10.74 |
-10.89 |
3 |
1-hexene | 1.73 |
2.19 |
15 |
hexanal | -2.85 |
-2.34 |
4 |
2,3-dimethyl-1,3-butadiene | 0.40 |
1.16 |
16 |
2-butanone | -3.76 |
-3.58 |
5 |
toluene | -0.77 |
-0.91 |
17 |
methylformate | -2.82 |
-4.01 |
6 |
tert-butylbenzene | -0.44 |
-0.11 |
18 |
ethylpropionate | -2.83 |
-2.80 |
7 |
dichloromethane | -1.42 |
-0.30 |
19 |
isoamylacetate | -2.24 |
-2.23 |
8 |
1,3-dibromopropane | -1.99 |
-3.01 |
20 |
propylamine | -4.56 |
-4.61 |
9 |
chloroethylene | 0.50 |
-0.39 |
21 |
dibutylamine | -3.38 |
-2.74 |
10 |
1,4-dichlorobenzene | -1.02 |
-1.06 |
22 |
1-nitropropane | -3.38 |
-2.64 |
11 |
diethyl sulfide | -1.45 |
-1.28 |
23 |
4-ethylpyridine | -4.66 |
-4.41 |
12 |
diisopropyl ether | -0.50 |
-1.87 |
24 |
2-isobytylpyrazine | -5.11 |
-3.82 |
3.4 讨论
由于有机化合物种类多、数量大、结构多样,如何用较少的特征参数描述分子,以建立与有机分子在水溶液中的溶解自由能的定量构效关系一直以来是难以解决的能量计算问题。本文所选的155个有机小分子化合物包括:烷烃、环烷烃、烯烃(又包括链烯烃、环烯烃、二烯烃)、炔烃、芳香烃、卤代烃、醇、醚、醛、酮、酸、脂,以及含有S、N元素的杂原子化合物、杂环化合物等,基本包括常见的有机小分子化合物。应用8个特征参数描述分子,用支持向量回归算法,通过核函数参数的优化,建立预测模型,为比较SVR法的预测能力,同时采用多元线性回归(MLR)方法建立了8个特征参数与溶解自由能的定量构效关系,相关结果见表2、表3,多元线性回归方程为:
y=-0.29158+1.722023LogP-0.09564TPSA-0.62209natoms-0.01801MW+0.947228Non-0.80409 NOH,NH+0.053023 Nrotb +0.0115V (3)
n=155 m=8 r2=0.80 s =1.12 F=73.6
n、m、 r2 、s、 F分别为回归方程的样本数、变量数、相关系数、估计标准误差和Fischer检验值。同时, 对方程所含的八个参数进行了相关分析, 结果表明这八个参数之间无自相关。比较可见,SVR法对预测样本具有较强的泛化能力,由图3可以看出所有样本都落在过原点450直线附近且无异常点,表明SVR法计算结果具有较好的稳定性。非线性拟合SVR算法建立的预测模型,拟合效果好;相比之下,线性回归方法只是把非线性关系近似地用线性方式处理,因此误差较大,相关系数偏小。从表1可见,以以上八个参数为输入向量,采用支持向量机方法进行回归建模,拟合效果好,能充分反映有机小分子结构与溶解自由能间的定量关系。
为了进一步将SVR算法结果与其他相关方法的拟合、预测结果[5,6] 相比较,表2、表3还分别列出了各种算法计算得到的相关系数(r2)、变量数(numvari)、最大偏差(maxdev)、最小偏差(mindev)、偏差大于1 kcal/mol(dev>1)样本的个数、最大实验值(maxexp)、最小实验值(minexp)、均方根误差(rms)、样本数(numsamp)等数值。其中,rms=
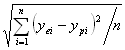 yei为实验值,ypi为支持向量机回归算法的拟合值或预测值,n为分析数据集的样本数。由表2、表3可见,支持向量机回归算法在数据的拟合和预测方面均优于其他方法,且所用参数相对较少。
yei为实验值,ypi为支持向量机回归算法的拟合值或预测值,n为分析数据集的样本数。由表2、表3可见,支持向量机回归算法在数据的拟合和预测方面均优于其他方法,且所用参数相对较少。
表2 训练集化合物计算溶解自由能( kcal/mol)的比较
| method | r2 | num vari | max dev | min dev | dev>1 | max exp | min exp | rms | num samp |
| MLR | 0.80 | 8 | 3.52 | 0.02 | 47 | 2.20 | -10.61 | 1.09 | 131 |
| SVR GA |
0.95 0.89 |
8 3 |
2.11 2.19 |
0.00 0.00 |
9 28 |
2.20 2.20 |
-10.61 -10.61 |
0.51 0.77 |
131 131 |
| HLOGS | 0.95 | 18 | 3.40 | 0.00 | 14 | 3.21 | -10.61 | 0.58 | 265 |
| ALOGS | 0.96 | 67 | 4.53 | 0.00 | 11 | 3.21 | -10.61 | 0.53 | 265 |
表3 预测集化合物计算溶解自由能( kcal/mol)的比较
method |
r2 |
num |
max |
min |
dev>1 |
max |
min |
rms |
num |
MLR |
0.80 |
8 |
2.21 |
0.04 |
9 |
2.56 |
-10.74 |
1.09 |
24 |
SVR |
0.94 |
8 |
1.37 |
0.01 |
5 |
2.56 |
-10.74 |
0.67 |
24 |
HLOGS |
0.86 |
18 |
2.47 |
0.04 |
8 |
2.90 |
-10.74 |
1.07 |
27 |
ALOGS |
0.92 |
67 |
2.47 |
0.01 |
5 |
2.90 |
-10.74 |
0.82 |
27 |
4 结论
[1] Cramer C J, Truhlar D G. Chem. ReV. ,1999, 99:2161~2200.
[2] Kollman P A. Acc. Chem. Res., 1996, 29: 461~469.
[3] Butina D, Gola J M R. J. Chem. Inf. Comput. Sci., 2003, 43,:837~841.
[4] Bruneau P. J. Chem. Inf. Comput. Sci., 2001, 41:1605~1616.
[5] Hongsuk K, Hwanho C, Hwangseo P. J. Chem. Inf. Model., 2007, 47: 509~514.
[6] Vellarkad N V, Arup K G, Chandra S U., et al. J. Chem. Inf. Comput. Sci., 1999, 39: 405~412.
[7] Vapnik V. Statistical Learning Theory. Wiley ,New York ,1998.
[8] Cortes C, Vapnik V. Machine Learning, 1995,20:273~297.
[9] Wan V, Campbell W M. Neura Networks for signal Processing―Proceeding of the IEEE workshop, Sydney,2000(2),:775~784.
[10] Thorsten J. Ph.D. Dissertation, Universitact Dortmund, 2001.
[11] Burbidge R, Trotter M, Buxton B, et al. Compu. Chem.2001,26(1):5~14.
[12] Zhou P, Zeng H,Li B,et al. Chemistry(Huaxue Tongbao),2006,5:342-347.
[13] Si H Z,Yao X J,Liu H X,et al. Acta Chimica Sinica(Huaxue Xuebao),2006,64(5):415-422.
[14] Zhang X D,Yu C,Guan H Y,et al. Journal of Shanxi University(Natural Science Edition)(Shanxi Daxue Xuebao(Ziran Kexue Ban)),2007,30(2):240-244.
[15] Molinspiration Cheminformatics, Bratislava, Slovak Republic.(http://www.molinspiration.com).
[16] Matheus P F. Org. Biomol. Chem., 2006, 4: 1154~1159.
[17] http://www.daylight.com/smiles/