http://www.chemistrymag.org/cji/2008/102009pe.htm |
Feb. 1,
2008 Vol.10 No.2 P.9 Copyright |
Liu Pengyan, Zhang Li, Liu Lei
(Department of Chemistry and Environment, Hebei University, Baoding 071002, Hebei, China)
Abstract An analytical method for analysing acrylamide in potato chips was validated. The acrylamide in potato chips was extracted with methanol, frozen, defatted with n-hexane, and purified with ENVI-18 solid-phase extraction (SPE) cartridge and then analyzed by HPLCĻCDAD for quantification. The limit of detection (LOD) was 11.5
mg/kg. Recoveries of acrylamide from samples spiked at levels of 500, 1000 and 2000 mg/kg (n = 3 for each level) ranged between 75.9 and 93.4% with relative standard deviations (RSD) of less than 8.4%. The results demonstrated that this method should be regarded as a new, low-cost, and robust alternative for conventional investigation of acrylamide.Keywords Acrylamide, Potato chips, HPLC
1. INTRODUCTION
Potato is one of the most popular crops in the world and it is consumed by millions of
people from different countries. In addition the potato chips taste so good that a great
number of people like it.
In April 2002 the Swedish National Food Administration reported the
discovery of relatively high levels of acrylamide in heat-treated potato products like
potato chips [1]. The discovery has attracted considerable interest and wide
attention from all over the world. The carbohydrate-rich foods seem to have relatively
more acrylamide. Many researchers have confirmed the existance of acrylamide in the foods
which are processed or cooked in different ways and it was announced that its
concentration might as high as several mg/kg [2-5].
Long-term exposure to acrylamide may cause damage to the nervous system
both in humans and animals to a certain extent [6,7], and acrylamide is also
considered as a potential genetic and reproductive toxin with mutagenic and carcinogenic
properties in experimental mammalians both in vitro and in vivo study [8-10].
So the development and validation of sensitive and reliable analytical methods for the
quantification of acrylamide in different food matrices was considered as essential.
At present, several
analytical methods are available for determining acrylamide in foods and the majority are
classical methods based on high performance liquid chromatography (HPLC) or gas
chromatography (GC) techniques [11-16]. But, analysis of acrylamide in food
products focuses primarily on chromatographic methods such as HPLCĻCMS [2,17-19]
The method utilizes HPLC with DAD detection that can be adopted by analytical laboratories easily. Some researchers have used this method [23], but we have differents with them in purification, SPE cartridge and so on. The acrylamide in potato chips was extracted with methanol, frozen, defatted with n-hexane, and purified with ENVI-18 solid-phase extraction (SPE) cartridge and then analyzed by HPLC-DAD for quantification. After these steps the extraction becomes clear. What's more, all the results indicated that it was a reliable method for the quantification of acrylamide in potato chips.
2. EXPERIMENT METHOD
2.1 Sample extration
The potato chips were pulverized and
homogenized. As for sample, 1 g of above-mentioned samples was weighted into a 10 mL glass
centrifuge tube with cap. The sample was spiked with acrylamide to achieve the final
concentrations of 500, 1000 and 2000 mg/kg to determine recovery at this stage. The sample was
suspended in 4.0 mL of methanol and extracted for 2 min in WH-861 variable speed vortex
shaker. The tubes were then clamped and shaken in a constant-temperature shaker for 20 min
and centrifuged at 4000 rpm for 10 min. The clear supernatant was transferred into another
10 mL glass centrifuge tube by a pipet. The residues were extracted again by 2 ĄÁ 3
The fluids were placed in a water bath at 40oC and evaporated to 3 mL under a gentle stream of nitrogen. The reaction mixture was placed in the refrigeratory for two hours at 4oC and centrifuged at 4000 rpm for 10 min. The clear supernatant was transferred.
To make a defatting process, 3 mL of n-hexane was added and each tube was then capped and shaken by hand or vortex briefly to mix the contents of tube. The tubes were centrifuged at 4000 rpm for 10 min. The fluids were extracted with 3 mL of n-hexane for three times. The supernatant was discarded and concentrated to 0.8 mL under a gentle stream of nitrogen.
2.2 Sample clean-up
For the SPE clean-up, ENVI-18 cartridge was preconditioned with 5 mL of methanol, 5 mL of water and 5 mL of methanol in turn at rate of one drop per four seconds. The methanol and water portions were discarded. Then, 1 ml of the extract was passed through the cartridge at the same rate using a syringe. Then the cartridge was eluted with 2 ĄÁ 1 mL of methanol and the eluant was collected. Then it was placed in a water bath at 4oC and evaporated to dryness under a gentle stream of nitrogen. The residue was immediately redissolved in 1 mL of 5% (v/v) methanol in water by mixing in a vortex mixer for 1 min and filtered through a 0.45 mm syringe filter. Then 10 mL of the final test solution was injected into HPLC column for quantification by HPLC-DAD.
3 RESULTS AND DISSCUSSION
3.1 Extraction Solvent
Extraction of acrylamide from the food is a critical step since acrylamide may be
firmly enclosed and not homogeneously distributed. In general, pure water enabled higher
acrylamide recovery compared to methanol. The use of pure water gives effective extraction
of acrylamide, so the sample preparation was usually started by extracting the ground
sample with water enough for a proper swelling in most of the methods based on HPLC
coupled to tandem MS detection system. [3, 5, 11, 24, 25] But coextraction of
other unwanted compounds in the matrix (e.g. pigments, carbohydrates, food addiitive)
might interfere with the detection and degenerate the chromatographic system. It is
difficult to obtain a clear supernatant by centrifugation.
On the other hand,
acrylamide is also highly soluble in methanol (155.0 g/100 mL) and much compatible with
the food matrices containing high amounts of fat such as potato chips, when methanol
In addition, in order to extract completely, the samples were extracted another two times. Extraction was considered complete at 20 min because of the second extract contained less than 10% of the acrylamide concentration of the first extract.
3.2 Defatting
After sample extraction, in order to obtain a clear supernatant, it was assayed an additional freeze-unfreeze cycle. [26] After low temperature (4oC) it was observed a cloudy supernatan. Finally the tubes were centrifuged at 4000 rpm for 10 min and a clear extraction was gotten.
The use of defatting with n-hexane had in most cases no significant effect on the acrylamide yield, not even for fatty matrices. Defatting was accomplished in the same tube as the extraction and the n-hexane was discarded throughout the whole extraction (giving a two-phase extraction step). The risk for loss of sample prior to extraction that could otherwise occur when the n-hexane was removed was thereby minimised. In order to evaluate the effectivity of n-hexane on the defatting of acrylamide in potato chips, a experimental design was used (Table 1). The experiment was performed with replicates. It showed, however, significance for the purification of potato chips, but the effect of loss was negligible.
Nevertheless, defatting might have other advantages, for example, prevention of clogging/overloading the SPE-columns and longer lifetime of the HPLC-columns.
Table 1 Effect of the defatting on acrylamide analysis
Concentrationi ( mg/mL) |
Before defatting (area) |
After defatting (area) |
0.5 |
35404 |
34959 |
0.8 |
56186 |
55364 |
1.0 |
70757 |
69240 |
3.3 Optimization of SPE Procedure
It was found necessary to purify acrylamide extracts by solid-phase extraction (SPE). Normal-phase (Florisil) and reversed-phase (ENVI-18) columns were tested for their ability to separate acrylamide and impurity.
The functionalization with hydroxyl groups of ENVI allows the retention of polar ionizable and/or non-ionizable analytes from aqueous samples, so it could be a good choice of column for the clean-up of acrylamide.
Because of the high polarity of acrylamide, methanol was chosen as eluting solvent for ENVI. However, methanol could offer some advantages such as easier solvent evaporation and consequent higher pre-concentration in ENVI. Nevertheless, a minimum elution volume was required in order to achieve a maximum enrichment factor with an evaporation step. To know how much eluting solvent was needed, the elution curve [recovery (%) versus volume of elute] was describe (Fig.1); from this curve, the minimum solvent volume needed to remove the retained acrylamide quantitatively was found to be 3 mL for the ENVI column.
Good absorbability and SPE recovery of acrylamide were found when using ENVI-18 SPE cartridges, i.e. 92.4% of SPE recovery under the acrylamide concentration of 2 mg/mL. It was showed that the method could be properly used for the clean-up of acrylamide in food samples.
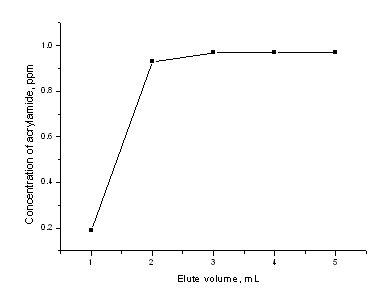
Fig. 1 The change of acrylamide concentrations in the eluate fractions collected during the elution through ENVI-18 cartridge. Sample loaded onto the cartridge was 1.0 mL of acrylamide working standard solution at a concentration of 2.0 mg/mL. The results in Figure 1 revealed that each millilitre of methanol was added to rinsed the cartridge and it was analyzed by HPLC. It could be observed no acrylamide presence after 2 mL methanol. To make sure there was no acrylamide in the effusion, another 1 mL methanol was added for acrylamide extraction.
The effect of flow rate values was also evaluated within the range 0.1ĻC1.0 mL/min. It was found that 0.8 mL/min was the optimum value.
On the other hand we study the Florisil for the chean-up. We produced the column ourselves (500 mg, 3 mL). Florisil cartridge was preconditioned consequently with 25% (v/v) acetone in n-hexane (6 mL) and followed by 50% (v/v) acetone in n-hexane (6 mL) at rate of one drop per four seconds. The mixtures were discarded. Then, 1 mL of acrylamide working standard solution at a concentration of 2.0 mg/mL was passed through the cartridge at the same rate using a syringe. Then the cartridge was eluted with 6 ĄÁ 1 mL of 50% (v/v) acetone in n-hexane and the eluant was collected. Then it was placed in a water bath at 40oC and evaporated to dryness under a gentle stream of nitrogen. The residue was immediately redissolved in 1 mL of 5% (v/v) methanol in water by mixing in a vortex mixer for 1min and filtered through a 0.45 mm syringe filter. Then 10 mL of the final test solution was injected into HPLC column for quantification by HPLC-DAD. To our surprise there was no acrylamide .Then eluting solvents were changed (e.g. 75% (v/v) acetone in n-hexane, methanol, and water). The same results were gotten. The reason for acrylamide losses is not clear. As everyone knows, the polarity of acrylamide is strong which is same as the Florisil. Maybe they formed close connection. And the connection was so strong that it couldn't be separated by those eluting solvents.
So ENVI-18 cartridge is a good choice of column for the clean-up of acrylamide comparing to the Florisil cartridge at this condition.
3.4 Spectral Analysis
Since acrylamide is a derivative of carboxylic acid, it has a maximum absorption within the wavelength range of 195-205 nm. However, all co-extractives from the food matrix also absorb well in this wavelength range adversely affecting the detection accuracy. On the other hand, acrylamide has also a good absorption at 210 nm (Fig.2). In addition it can be analyzed at 210 nm in complex food matrices like potato chips with more precision and accuracy.
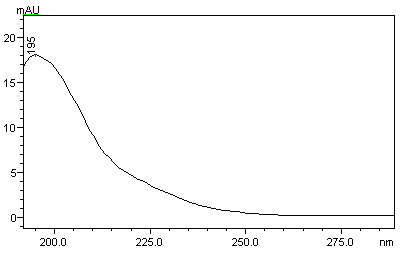
Fig. 2 UV absorbance spectra of acrylamide recorded (peak maximum at 210 nm).
3.5 Validation of the Acrylamide Analysis
VP-ODS column was assayed, founding excellent acrylamide chromatogram (retention times ranged from 6 to 8 min) (Fig.3).
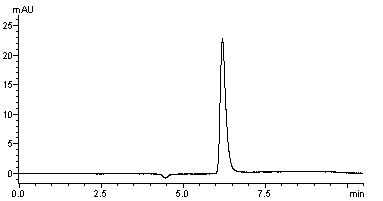
Fig. 3 Chromatogram of the acrylamide at a level of 5 mg/mL.
Stock solution of standards (1.0 mg/mL) and working
standard solutions (5.0, 2.0, 1.0, 0.5 and 0.1 mg/mL) were prepared in methanol. They were injected onto HPLC
column for quantification by HPLCĻCDAD.
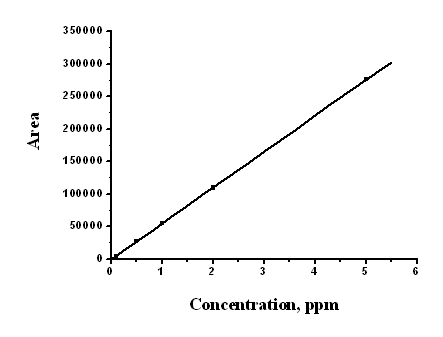
Fig. 4 Calibration standards
curve of acrylamide.
A good coefficient of correlation of 0.99999 was achieved
over the whole concentration range 0.1-5 mg/mL (y = 55287.3429 x - 714.42979) (Fig.4).
For quantitative
purposes, the method was validated by defining the repeatability, precision, and recovery.
The repeatability of the method was estimated for determination of acrylamide from potato
crisps.
The precision of the method was determined by calculating the relative
standard deviations (RSD) of the replicate measurements. Table 2 showed the results of a
day precision study in which portions of three levels of solution of standards were
repeatedly analyzed five times for the inter-day precision study.
Table 2 Analysis of the precision (n=5)
concentration(mg/L) |
0.5 |
1.0 |
2.0 |
Area |
37368 |
56407 |
124110 |
Mean area |
37267.5 |
59148.2 |
126481 |
RSD (%) |
2.3 |
3.7 |
1.5 |
Additionally,
acrylamide concentrations of 20 mg/kg could be successfully detected in potato chips. The
chromatograms were displayed in Figure 5 and Figure 6 respectively.
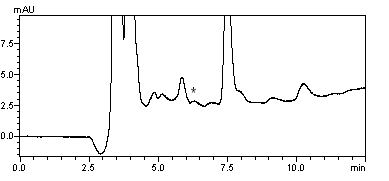
Fig. 5 Chromatograms of
acrylamide in potato chips.
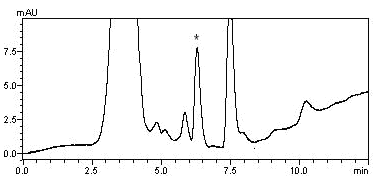
Fig. 6 Chromatogram of potato chips spiked
with acrylamide at a level of 2000 mg/kg.
Accuracy
for acrylamide by LC-DAD was excellent
with the values for the RSD of each data set ranged between 4.3 and 8.4% (Table 3).
Recovery rates were
demonstrated in three tests employing the method of standard addition ranging from 500 to
2000 mg/kg. Good recoveries
were achieved for the validated HPLC-DAD method,
confirming the reliability of the methods. The mean percentage recoveries exceeded 75% for
all spiking levels for samples (Table 3). Three measurements of the certified reference
material averaged to an acrylamide concentration of 36 mg/kg.
Table 3 Recovery assay from potato chip reference material spiked with
acrylamide
Acrylamide added (mg/kg) |
Recovery (%) |
Mean recovery(%) |
RSD |
||
1 |
2 |
3 |
|||
500 |
|
78.6 |
93.4 |
|
8.4 |
1000 |
|
75.9 |
82.7 |
|
4.3 |
2000 |
|
89.9 |
91.8 |
|
4.6 |
Limit of detection (LOD) and limit of quantification (LOQ) were
determined as the amount of analyte that produced a signal to noise ratio of 3:1 and 10:1,
respectively. LOD and LOQ were calculated using a blank sample spiked with very low
amounts of acrylamide in the beginning of the extraction. In the present study, the limits
of detection and quantification of acrylamide were 11.5 and 38.3 mg/kg, respectively.
Considering all the
above data for method validation test, the LCĻCDAD
method established in the present study and sample pretreatment procedures employed in the
present work can be regarded as selective, precise, and robust.
REFERENCES
[1] Swedish National Food Administration: Information about Acrylamide in Food, http://www.slv.se/Download/Document/approvedDocs/enginformationakryl.htm.
[2] Rosén J, Hellenäs K E. Analyst, 2002, 127: 880.
[3] Eden T, Per R, Patrik K, et al. J. Agric. Food Chem., 2002, 50: 4998.
[4] Ahn J S, Castle L, Clark D B, et al. Food Addit. Contam., 2002, 19: 1116.
[5] Adam B, Benjamin P-Y L, David L, et al. J. Agric. Food Chem, 2003, 51: 802.
[6] Tilson H A. Neurobehav. Toxicol. Teratol., 1981, 3: 445.
[7] LoPachin R M, Lehning E J. Neurotoxicology, 1994, 15: 247.
[8] Kerry L D, Charles O A, Myron S O, et al. J.. Mutat. Res., 1988, 195: 45.
[9] Costa L G, Deng H, Gregotti C, et al. Neurotoxicology, 1992, 13: 219.
[10] Kerry L D, George R D, Udo H E, et al. Mutat. Res., 1995, 330: 71.
[11] Alain P, Adrienne P, Jean-Marie O. J. Chromatogr. A, 2004, 1035: 123.
[12] Denis A, John A G R, Martha L G, et al. J. Agric. Food Chem., 2004, 52: 1996.
[13] David S B, Jason H, Richard M L, et al. J. Chromatogr. B, 2001, 758: 289.
[14] Bologna L S, Andrews F F, Barvenik F W, et al. J. Chromatogr. Sci., 1999, 37: 240.
[15] Laurence C. J. Agric. Food Chem., 1993, 41: 1261.
[16] Magnus J, Peter S. J. Agric. Food Chem., 2003, 51: 7866.
[17] Kuniaki K, Tsuyoshi I, Akiko T et al. J. Chromatogr. A, 2001, 911: 75.
[18] Bermudo E, Moyano E, Puignou L, et al. Analytica Chimica Acta, 2006, 559: 207.
[19] Ono H, Chuda Y, Ohnishi-Kameyama M et al. Food Addit. Contam., 2003, 20: 215.
[20] Biedermann M, Biedermann-Brem S, Noti A, et al. Mitt. Lebensm. Hyg., 2002, 93: 638.
[21] Christian G, Sabine K, Eur. J. Lipid Sci. Technol., 2002, 104: 762.
[22] Nemoto S, Takatsuki S, Sasaki K, J. Food Hyg. Soc. Jpn., 2002, 43: 371.
[23] Vural G, Hamide Z S, Jale A, J. Chromatogr. A, 2005, 1088: 193.
[24] Croft M, Tong P, Fuentes D, et al. Food Addit. Contam., 2004, 21: 721.
[25] Calbiani F, Careri M, Elviri L, et al. J. AOAC Int., 2004, 87: 107.
[26] Sonja R, Richard H S, J. Chromatogr. A, 2003, 1020: 121.
ÁõÆMŅŌĢŽÕÅĀöĢŽÁõĀÚ
(šÓąąīóŅ§ŧŊŅ§Óëŧ·ūģŋÆŅ§Ņ§ÔšĢŽšÓąąĢŽąĢķĻĢŽ071002)
ÕŠŌŠ ąūÎÄēÉÓÃķþžŦđÜÕóÁОėēâÆũ-ļßЧŌšÏāÉŦÆŨēâķĻĘíÆŽÖÐĩÄąûÏĐõĢ°·ĢŽēÉÓÞŨīžĖáČĄąûÏĐõĢ°·ĢŽĩÍÎÂīĶĀíšÍÕýžšÍéÁŠšÏČĨģýÓÍÖŽž°ÔÓÖĘĢŽC18đĖÏāÝÍČĄÐĄÖųūŧŧŊĖáČĄŌšĢŽŌÔķþžŦđÜÕóÁОėēâÆũēâķĻÆ䚎ÁŋĄĢ·―·ĻŧØĘÕÂĘÔÚ75.9%-90.9%ÖŪžäĢŽžėēâÏÞΊ11.5mg/kgĢŽÏāķÔąęŨžÆŦēîÐĄÓÚ8.4%ĄĢČĨģýÓÍÖŽšÍļÉČÅÔÓÖĘĘĮĘíÆŽŅųÆ·īĶĀíÖÐĩÄđØžüŧ·―ÚĢŽķÔÓÚąĢÖĪĘĩŅéĩÄģÉđĶūßÓÐÖØŌŠŌâŌåĢŽļũđúŋÆŅ§žŌÖÂÁĶÓÚĘíÆŽŅųÆ·ūŧŧŊĩÄŅÐūŋĢŽąūēÉÓÃĩÍÎÂīĶĀíšÍÕýžšÍéÁŠšÏČĨģýÓÍÖŽž°ÔÓÖĘūŧŧŊЧđûšÃĢŽ―ĩĩÍÁËļÉČÅĢŽžōąãÓÐЧĢŽŋÉŨũΊŅųÆ·ūŧŧŊīĶĀíĩÄ·―·ĻĄĢ
đØžüīĘ ąûÏĐõĢ°·ĢŧĘíÆŽĢŧļßЧŌšÏāÉŦÆŨ·Ļ ĄĄ