http://www.chemistrymag.org/cji/2008/104018pc.htm |
Apr. 1,
2008 Vol.10 No.4 P.18 Copyright |
(College of Materials Science and Engineering, Huaqiao University, Quanzhou 362021, China)
Abstract
Cloud point extraction of trace Pyrene from aqueous solution with sodium dodecyl sulfate (SDS) which determined by HPLC was carried out. The parameters affecting the separation and detection were investigated, which include SDS, HCl, NaCl, extraction time and temperatureŁ®Under the optimized condition, the concentrations of Pyrene showed a linear range of 0-10.0 mg·L-1 with R2=0.9994. The spiked recoveries and the detection limit of the proposed method were found to be 80.7-105.1% and 13.6 mg·L-1, respectively. The experimental results indicated that the proposed method was successfully applied to the determination of Pyrene in real water samples.Keywords Cloud point extraction(CPE)Ł»PyreneŁ»Sodium dodecyl sulfate(SDS)Ł»High performance liquid chromatography (HPLC) ×ǵăÝÍȡ-¸ßЧҺĎŕÉ«ĆײⶨˮÖĐşŰÁżµÄÜĹ
łÂşçŔö
ѦĐăÁá Đěç⣨»ŞÇČ´óѧ˛ÄÁĎżĆѧÓ빤łĚѧԺŁ¬¸Ł˝¨ ČŞÖÝ362021Ł© 2008Äę1ÔÂ14ČŐĘŐ¸ĺ;¸Ł˝¨ĘˇÇŕÄęČ˲Ŵ´Đ»ů˝đŁ¨No.2005J029Ł©ˇ˘ąúÎńÔşÇČ°ěżĆŃĐ»ů˝đŁ¨No.05QJR05Ł©ˇ˘»ŞÇČ´óѧ¸ß˛ă´ÎŇý˝řČ˲ſĆŃĐ»ů˝đ×ĘÖúĎîÄż
ŐŞŇŞˇˇ˛ÉÓĂŇőŔë×Ó±íĂć»îĐÔĽÁĘ®¶ţÍé»ůÁňËáÄĆ(sodium dodecyl sulfate, SDS)×ǵăÝÍȡ(Cloud Point Extraction,CPE)Ł¬¸ßЧҺĎŕÉ«Ć×(High performance liquid chromatography,HPLC)˛â¶¨Ë®ÖĐşŰÁżµÄÜĹ(Pyrene)Ł¬żĽ˛ěĘĘŇ˵ķ´Ó¦ĚőĽţşÍÓ°ĎěŇňËŘ(SDSˇ˘HClşÍNaCl)ˇŁÔÚ×îĽŃĚőĽţĎÂŁ¬·˝·¨Ľě˛âĎŢÎŞ13.6 mg·L-1Ł¬ĎßĐÔ·¶Î§ÎŞ0ˇ«10.0 mg·L-1Ł¬»ŘĘŐÂĘÎŞ80.7ˇ«105.1%ˇŁ·˝·¨żěË١˘Ľň±ăˇ˘ľĽĂŁ¬Ó¦ÓĂÓÚË®ÖĐşŰÁżÜŵIJⶨŁ¬˝áąűÂúŇ⡣
ąŘĽü´Ęˇˇ×ǵăÝÍȡŁ»ÜĹŁ»Ę®¶ţÍé»ůÁňËáÄĆŁ»¸ßЧҺĎŕÉ«Ć×
¶ŕ»··ĽĚţ(polycyclic aromatic
hydrocarbons, PAHs)ĘÇąă·ş´ćÔÚÓÚ»·ľłÖеÄÓжľÓĐ»úÎŰČľÎĆ仯ѧĐÔÖĘÎȶ¨Ł¬ÔÚ»·ľłÖĐł¤ĆÚÁô´ćŁ¬˛˘Í¨ąý»·ľłĐî»ýˇ˘ÉúÎďĐî»ýˇ˘ÉúÎďת»Ż»ň»ŻŃ§·´Ó¦µČ·˝Ę˝ÎŰČľ»·ľłŁ¬ÍţвÉúÎ。żµŁ¬ŇŃłÉÎŞČ«Çň»·ľłÎŰČľŃĐľżµÄÖŘŇŞÄÚČÝşÍČȵăżÎĚâ[1]ˇŁ
×ǵăÝÍȡ(Cloud Point ExtractionŁ¬CPE)ĘÇ˝üÄęŔ´łöĎÖµÄŇ»ÖÖĐÂĐÍŇşŁŇşÝÍȡĽĽĘőŁ¬ËüŇÔ±íĂć»îĐÔĽÁ˝şĘřË®ČÜŇşµÄČÜ˝âĐÔşÍ×ǵăĎÖĎóÎŞ»ů´ˇŁ¬Í¨ąý¸Ä±äʵŃéĚőĽţ(Čçζȡ˘µç˝âÖʵČ)Ňý·˘Ë®ĎŕşÍ±íĂć»îĐÔĽÁĎŕ·ÖŔ룬¸Ă·¨˛»ĘąÓûӷ˘ĐÔÓĐ»úČÜĽÁŁ¬ľßÓĐÝÍȡÂʸߡ˘¸»ĽŻ±¶Ęý´óŁ¬˛Ů×÷Ľň±ăˇ˘°˛Č«Ł¬żÉʵĎÖÁŞÓĂ»ŻµČÓŵ㣬Ňň´ËĘܵ˝ČËĂǵļ«´óąŘעŁ¬ÔÚÉúÎď´ó·Ö×Ó·ÖŔë´ż»Żˇ˘ÓĐ»úС·Ö×Ó·ÖŔë˛â¶¨Ľ°˝đĘôŔë×Ó·ÖŔ븻ĽŻµČ·˝ĂćµĂµ˝ąă·şÓ¦ÓĂ[2-4]ˇŁ
ÓĂÓÚ×ǵăÝÍȡPAHsµÄ¶ŕÎŞ·ÇŔë×ÓĐͱíĂć»îĐÔĽÁŁ¬ČçTriton X ϵÁĐ[5-8], PONPE ϵÁĐ[9,10]şÍBrijϵÁĐ[11-14]]µČŁ¬¶řŇőŔë×ÓĐͱíĂć»îĐÔĽÁSDSÓĂÓÚ×ǵăÝÍȡÓĐŇÔĎÂÓŵ㣺ÝÍȡЧąűşĂŁ¬Ň×ÓÚ·ÖŔ룬ÇҵͶľŁ¬ĽŰÁ®Ł¬±ľŃĐľżĘąÓĂHClĘąSDSÖĘ×Ó»ŻŁ¬ĽÓČëNaCl˝µµÍĚĺϵµÄ×ǵ㣬¸ßЧҺĎŕÉ«ĆײⶨŁ¬ĚÖÂŰÓ°Ďě×ǵăÝÍȡşŰÁżÜŵĸ÷ĎîŇňËŘŁ¬˝¨Á˘ÁËË®ÖĐşŰÁżÜĹĽňµĄˇ˘żěË١˘ÁéĂô˛â¶¨µÄĐ·˝·¨ˇŁ
1ˇˇĘµŃ鲿·Ö
¸ßЧҺĎŕÉ«Ć×ŇÇ(Agilent1100, ĂŔąú°˛˝ÝÂ׿ƼĽÓĐĎŢą«Ëľ)Ł»UV-4802HĐÍ×ĎÍâżÉĽű·Öąâąâ¶ČĽĆ(ÓČÄáżÂ(ÉĎşŁ)ŇÇĆ÷ÓĐĎŢą«Ëľ)Ł»KQ-100ĐÍł¬Éů˛¨ÇĺĎ´Ć÷(ŔĄĂ÷ĘĐł¬ÉůŇÇĆ÷ÓĐĎŢą«Ëľ)Ł»DK-S24µçČČşăÎÂˮԡąř(ÉĎşŁľ«ĂÜʵŃéÉ豸ÓĐĎŢą«Ëľ)Ł»800BŔëĐÄ»ú(ÉĎşŁ°˛Í¤żĆѧŇÇĆ÷ł§)Ł»SHZ-D(˘ó)Ń»·Ë®Ę˝ŐćżŐ±Ă(ÉĎşŁşŘµÂʵŃéÉ豸ÓĐĎŢą«Ëľ)ˇŁ
ÜĹ(pyrene, Aldrich Chem.Co. , AR)Ł»Ľ×´Ľ(TEDIA COMPANY, INC.USA , HPLC)Ł»Ľ×´Ľ(ąă¶«ÉÇÍ·ĘĐÎ÷¤»Żą¤ł§, AR)Ł»Ĺ¨ŃÎËá(ÉĎşŁ»ŻŃ§ĘÔĽÁÓĐĎŢą«ËľŁ¬AR)Ł»SDS(Biosharp , AR)Ł»ÂČ»ŻÄĆ(ąă¶«ÉÇÍ·ĘĐÎ÷¤»Żą¤ł§, AR)ˇŁ
1.2ˇˇĘµŃé·˝·¨
1.2.1ÜĹ-Ľ×´Ľ×ĎÍâ×î´óÎüĘŐ˛¨ł¤µÄ˛â¶¨
·Ö±đȡ0ˇ˘0.05ˇ˘0.15ˇ˘0.3ˇ˘0.6ˇ˘1.0mL 10 mg·L-1ÜĹ-Ľ×´Ľ±ę׼ҺŁ¬ÓÚ10mLľßČűĘÔąÜÖĐŁ¬Ľ×´Ľ¶¨ČÝÖÁ5 mLŁ¬ŇˇÔȡŁŇԼ״ĽÎŞ˛Î±ČŁ¬ÓĂUV-4802HĐÍ×ĎÍâżÉĽű·Öąâąâ¶ČĽĆŁ¬´Ó200nmˇ«500nm¶ÔĽ×´ĽşÍÜĹ-Ľ×´Ľ±ę׼Һ˝řĐĐĆ×ͼɨĂ裬ȷ¶¨ÜĹ-Ľ×´ĽµÄ×î´óÎüĘŐ˛¨ł¤ˇŁ
1.2.2 ×ǵăÝÍȡąýłĚşÍ·ÖÎöÖ¸±ęµÄ˛â¶¨
ȡĘĘÁżÜĹ-Ľ×´Ľ±ę׼ҺÓÚ10 mLľßČűĘÔąÜÖĐŁ¬ŇŔ´ÎĽÓČëSDSˇ˘Ĺ¨HClˇ˘±ĄşÍNaClČÜŇşŁ¬ Milli-QË®¶¨ČÝÖÁ10 mLŁ¬ŇˇÔČŁ¬2000 rpmŔëĐÄ10 minŁ¬Ęą±íĂć»îĐÔĽÁĎŕÓëË®Ďŕ·ÖŔ룬¸»ĽŻÓÚÉϲ㣬ÓĂ×ÔÖĆȡŃůĆ÷ȡłöSDSĎ࣬Ľ×´Ľ¶¨ČÝÖÁ3 mLşóŁ¬¸ßЧҺĎŕÉ«Ć×ŇDzⶨˇŁÍ¬Ę±×öŇ»¸ůČ«łĚżŐ°×ąÜŁ¬Ľ´ĘÔąÜÖв»ĽÓÜĹ-Ľ×´ĽµÄ±ę׼ҺŁ¬ĆäËű˛Ů×÷ÍęȫͬÉĎŁ¬ŇÔĎűłýĘÔĽÁÎó˛îˇŁ
ÝÍȡÂʲÉÓñę׼ÇúĎß·¨˛â¶¨Ł¬ĎŕĚĺ»ý±ČŁ¬ĽňłĆĎŕ±ČŁ¬Ľ´·ÖŔ븻ĽŻşó±íĂć»îĐÔĽÁ¸»ĽŻĎŕ(Vs)şÍË®Ďŕ(Vw)µÄĚĺ»ý±ČˇŁ
1.2.3 ¸ßЧҺĎŕÉ«Ć×·ÖÎöĚőĽţ
É«Ć×ÖůŁşAichrom ODS-C18Öů(250 mmˇÁ4.0 mmŁ¬5 mm)Ł»Á÷¶ŻĎࣺĽ×´Ľ-Ë® (90Łş10)Ł»Á÷ËŮŁş0.8 mL·min-1Ł»ÖůÎÂŁş30 ˇćŁ»×ĎÍâÎüĘŐĽě˛âĆ÷˛¨ł¤Łş239 nmŁ»˝řŃůÁżŁş20 mLˇŁ
1.2.4 ŃůĆ·´¦Ŕí
×ÔŔ´Ë®ˇ˘ĐŁÔ°ÇďÖĐşţË®ˇ˘ČŞÖݶ«şţË®ÓĂ0.45 mmż×ľ¶ÂËĤąýÂËŇÔłýČĄË®ÖĐĐü¸ˇżĹÁŁÎÖĂÓÚ4 ˇć±ůĎäÖĐ´ýÓáŁ
2 ˝áąűÓëĚÖÂŰ
ÜĹ-Ľ×´Ľ±ę׼ҺµÄ×ĎÍâ-żÉĽűÎüĘŐąâĆ×ÍĽČçÍĽ1ËůĘľŁ¬ĎŕӦŨ¶Č·Ö±đÎŞ0ˇ˘0.1ˇ˘0.3ˇ˘0.6ˇ˘1.2ˇ˘2.0 mg·L-1ˇŁ
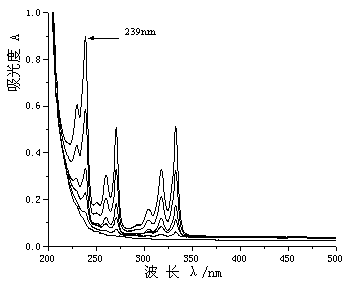
ÍĽ1 ÜĹ-Ľ×´Ľ±ę׼ҺµÄÎüĘŐąâĆ×ÍĽ
Fig. 1 UV-Vis absorption spectra of pyrene titer
ÓÉÍĽ1Ł¬ÜĹ-Ľ×´Ľ±ę׼ҺÔÚ200 nmˇ«500 nmÄÚÓжŕ¸öÎüĘշ壬×î´óÎüĘŐ·ĺÔÚ239 nm´¦Ł¬ËćÜĹŨ¶ČµÄµÝÔöŁ¬ĆäÎüąâÖµĎßĐÔÔöĽÓˇŁŇň´ËѡÔń239 nm×÷ÎŞ±ľĘµŃéĚĺϵµÄ˛â¶¨˛¨ł¤ˇŁ
2.2Ó°Ďě×ǵăÝÍȡµÄŇňËŘ
2.2.1 SDS¶Ô×ǵăÝÍȡµÄÓ°Ďě
SDSµÄŨ¶Čľö¶¨Á˱íĂć»îĐÔĽÁ¸»ĽŻĎŕµÄĚĺ»ýŁ¬˝ř¶řÓ°ĎěÝÍȡÂʺͷ˝·¨Ľě˛âĎޡŁÓÉÍĽ2Ł¬Ëć×ĹSDSµÄÔöĽÓŁ¬Ďŕ±ČÔöĽÓŁ¬ÜŵÄÝÍȡÂĘłĘĎÖĎČÔöĽÓşó˝µµÍµÄÇ÷ĘơŁŐâĘÇŇňÎŞSDSŨ¶ČąýµÍŁ¨ŁĽ1.25%ʱŁ©Ł¬ÝÍȡ˛»ÍęČ«Ł¬SDSŨ¶Čąý´óŁ¨Łľ2%ʱŁ©Ł¬±íĂć»îĐÔĽÁ·Ö×ÓĹöײ˝áşĎĽ¸ÂĘÔö´óŁ¬ČÝŇ׾ۼŻłÉŇşµÎŁ¬ÝÍȡÂĘ˝µµÍŁ¬Í¬Ę±ÔöĽÓÁËĎŕ±ČŁ¬˝µµÍÁ˸»ĽŻ±¶ĘýˇŁÁíÍâʵŃé·˘ĎÖŁ¬Ëć×ĹSDSŨ¶ČÔöĽÓŁ¬×ǵ㽵µÍŁ¬µ±SDSŨ¶ČÎŞ1.25ŁĄĘ±Ł¬×ǵăŇŃ˝µÖÁĘŇÎÂŁ¬ÔÚ±ŁÖ¤ÝÍȡÍęČ«µÄÇéżöĎÂŁ¬ĘµŃéѡÔńSDSŨ¶ČÎŞ2.0ŁĄˇŁ
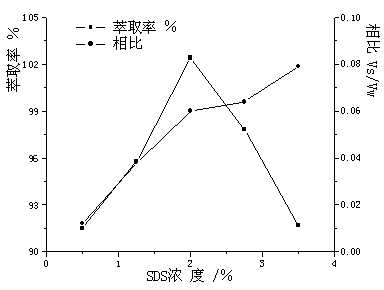
ÍĽ2 SDS¶ÔÝÍȡÂĘşÍĎŕ±ČµÄÓ°Ďě
Fig.2 Effect of SDS on
extraction efficiency and the phase volume ratio
עŁşPyrene=0.9 mg·L-1Ł¬HCl=3.0
mol·L-1Ł¬NaCl=0.92 mol·L-1
2.2.2 ŨHClˇ˘±ĄşÍNaCl¶Ô×ǵăÝÍȡµÄÓ°Ďě
ʵŃéÖĐĽÓČëHClµÄÄżµÄŁ¬ĘÇĘąSDSÖĘ×Ó»ŻŁ¬ÓÉŇőŔë×ÓĐÍת»ŻÎŞ·ÇŔë×ÓĐͱíĂć»îĐÔĽÁŁ¬˝şĘřŨ¶ČÔö¸ßŁ¬łöĎÖ»ë×ÇŁ¬×îÖŐĐÎłÉË«ŇşĎŕʵĎÖÓëË®ĎŕµÄ·ÖŔë[15-19]ˇŁNaClżÉ˝µµÍĚĺϵµÄ×ǵă,´Ů˝řĎŕ·ÖŔëµÄ·˘ÉúˇŁÍĽ3şÍÍĽ4ĎÔĘľÁËHClşÍNaCl¶ÔÝÍȡÂĘşÍĎŕĚĺ»ý±ČµÄÓ°Ď졣
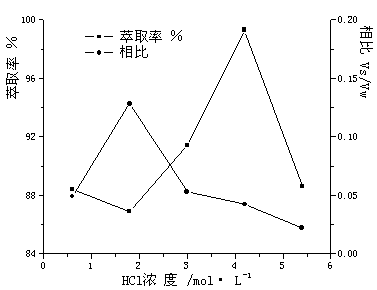
ÍĽ3 HCl¶ÔÝÍȡÂĘşÍĎŕ±ČµÄµÄÓ°Ďě
Fig.3ˇˇEffect of HCl on extraction efficiency and the phase
volume ratio
עŁşPyrene=0.9 mg·L-1Ł¬SDS=2%Ł¬NaCl=0.92 mol·L-1
ÓÉÍĽ3żÉÖŞŁ¬˝ĎµÍŨ¶ČµÄHClĘÇÎŞÁËČÜ˝âSDSŁ¬ĚĺϵÎŢ×ǵăĎÖĎóŁ¬HClŨ¶ČÔöĽÓŁ¬ÝÍȡÂĘÖđ˝ĄÔö´óŁ¬¶řąý¶ŕµÄ[H+]Ҳ˛»»áÔöĽÓÜŵÄÝÍȡÂʡŁ˝áąű±íĂ÷HClŨ¶ČÔÚ0.5ˇ«5 mol·L-1ʱľůżÉŇý·˘Ďŕ·ÖŔ롣ÇŇËćHClŨ¶ČµÄÔö´óŁ¬×ǵăÖ𽥽µµÍŁ¬ĎŕĚĺ»ý±ČÖ𽥼őСŁ¬Ĺ¨¶ČÎŞ4.2 mol·L-1ʱŁ¬×ǵ㽵ÖÁĘŇÎÂŁ¬ÝÍȡÂĘ×î¸ßŁ¬Í¬Ę±ĎŕĚĺ»ý±Č˝ĎСŁ¬Ĺ¨Ëő±¶Ęý˝Ď¸ßŁ¬Ňň´ËʵŃéѡÔńHClŨ¶ČÎŞ4.2 mol·L-1ˇŁ
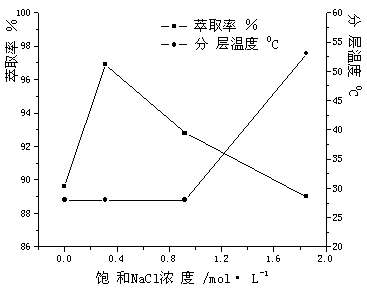
ÍĽ4 NaCl¶ÔÝÍȡÂʺͷÖĎŕµÄÓ°Ďě
Fig.4 Effect of NaCl on
extraction efficiency and the phase separation
עŁşPyrene=0.9 mg·L-1Ł¬SDS=2%Ł¬HCl=4.2 mol·L-1
ÍĽ4±íĂ÷ĚĺϵÖĐĽÓČëNaClŁ¬ÔÚĘŇÎÂĽ´ÓĐĂ÷ĎÔµÄ×ǵăĎÖĎóŁ¬Ĺ¨¶ČÎŞ0.31 mol·L-1ʱÝÍȡÂʿɴﵽ97%Ł¬±íĂ÷NaClµÄŃÎÎö×÷ÓĂŁ¬Ęą˝şĘřÖĐÇâĽü¶ĎÁŃŁ¬˝şĘřĽäłâÁ¦ĽőСŁ¬×ǵ㽵µÍŁ¬´ďµ˝±íĂć»îĐÔĽÁĎŕÓëË®ĎŕµÄ·ÖŔ롣
NaClŨ¶Č>0.31 mol·L-1ʱŁ¬ĚĺϵĂܶȼőСŁ¬Ë®ĎŕÓë±íĂć»îĐÔĽÁĎŕ·Ö˛ăËůĐčÄÜÁżĽÓ´óŁ¬ĐčŇŞĽÓČȵ˝53
ˇćŁ¬˛ĹżÉ´ďµ˝Á˝Ďŕ·ÖŔ룬ÔÚͬŃůÝÍȡĚőĽţĎÂŁ¬ÝÍȡÂĘ˝µµÍˇŁÁíÍ⣬ąýÁżµÄNaClŁ¨1.90 mol·L-1Ł©¸Ä±äÁËĚĺϵÖеÄŔë×ÓÇż¶ČŁ¬ÔěłÉSDS˛»Čܽ⣬ĚĺϵłöĎÖ°×É«Đő×´ÎÄŃÓëË®Ďŕ·ÖŔ룬µĽÖÂÁËÝÍȡÂĘĎ½µˇŁŇň´ËʵŃéѡÔńNaClŨ¶ČÎŞ0.31 mol·L-1ˇŁ
2.2.3 Ć˝şâζȺÍĆ˝şâʱĽä¶Ô×ǵăÝÍȡµÄÓ°Ďě
ËáĐÔĚőĽţĎÂŁ¬ĚĺϵÔÚĘŇÎÂĎÂĽ´żÉʵĎÖĎŕ·ÖŔ룬µ«şÄʱł¤Ł¬ÔĽ24hŁ¬Ë®ÔˇĽÓČČŁ¬żÉʹʱĽäËő¶ĚŁ¬ËµĂ÷ĽÓČȿɴٽřĎŕ·ÖŔ롣
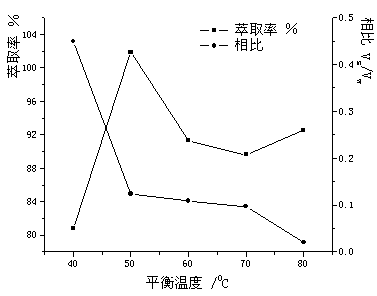
¸ůľÝʵŃ飬ƽşâʱĽäÔĽ30minĽ´żÉ´ďµ˝˝ĎşĂµÄÝÍȡЧąűŁ¬ĘµŃ鿼˛ěˮԡşăÎÂ30minŁ¬Ć˝şâζȶÔÜĹÝÍȡÂĘşÍĎŕ±ČµÄÓ°Ď죨ĽűÍĽ5Ł©ˇŁ
ÓÉÍĽ5żÉÖŞŁ¬ËćζČÉý¸ßŁ¬ÝÍȡÂĘѸËŮĚá¸ßŁ¬ÔÚ50ˇćʱ´ďµ˝×î¸ßŁ¬¶řşó˝µµÍŁ¬Ç÷ÓÚĆ˝şâˇŁĚĺϵζȽϵÍʱŁ¬SDSµÄÇ×Ë®»ůÍĹÓëË®·Ö×ÓĐÎłÉÇâĽüŁ¬PAHs˛»Ň×˝řČëSDS¸»ĽŻĎࣻζČÉý¸ßşóŁ¬ÇâĽü¶ĎÁŃŁ¬Ë®»Ż˛ă±»ĆĆ»µŁ¬SDSĘčË®ĐÔÔöÇżŁ¬Á˝Ďŕ·ÖŔë[9]ˇŁÍ¬Ę±ČČČŶŻÔöÇżŁ¬˝şĘřµÄÔöČÜżŐĽäÔö´óŁ¬Ë®Ďŕ˛ĐÁôµÄSDSŨ¶ČѸËŮĽőСŁ¬ÓĐŔűÓÚÝÍȡÂʵÄĚá¸ßˇŁµ«ÓÉÓÚČÜÖĘÔÚ˝şĘřÖеÄÔöČÜąýłĚÎŞ·ĹČČąýłĚŁ¬Éý¸ßζČĘą˝şĘř¶ÔČÜÖʵÄÔöČÜÄÜÁ¦ĽőČőŁ¬ÝÍȡÂĘ·´¶ř˝µµÍˇŁÔÚ40ˇ«80 ˇć·¶Î§ÄÚŁ¬Î¶ČÉý¸ßŁ¬±íĂć»îĐÔĽÁ¸»ĽŻĎŕÄýľŰЧąűÔ˝şĂŁ¬ĎŕĚĺ»ý±Č˝µµÍˇŁ×ۺϿĽÂÇŁ¬ĘµŃéѡÔń50
ˇćˇŁ
×ǵăÝÍȡĐčŇŞŇ»¶¨Ę±ĽäĘąSDSĐγɽşĘřŔ´°üąüĘčË®ÎďÖĘPAHsŁ¬˝«ĆäÝÍȡ˝řÄýľŰĎ࣬ÔÚ50
ˇćĎÂŁ¬ĘµŃ鿼˛ěÁËşăÎÂʱĽä¶ÔÝÍȡЧąűµÄÓ°Ď죬˝áąűČçÍĽ6ËůĘľˇŁ
ÍĽ5 Ć˝şâζȶÔÝÍȡÂĘşÍĎŕ±ČµÄÓ°Ďě
Fig.5 Effect of temperature on
extraction efficiency and the phase volume ratio
עŁşPyrene=0.9 mg·L-1Ł¬SDS=2%Ł¬HCl=4.2 mol·L-1Ł¬NaCl=0.31 mol·L-1
ÍĽ6 şăÎÂʱĽä¶ÔÝÍȡÂĘşÍĎŕ±ČµÄÓ°Ďě
Fig.6 Effect of time on extraction
efficiency and the phase volume ratio
עŁşPyrene=0.9 mg·L-1Ł¬SDS=2%Ł¬HCl=4.2 mol·L-1Ł¬NaCl=0.31 mol·L-1
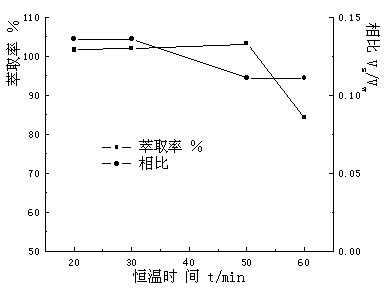
ÓÉÍĽ6żÉÖŞŁ¬şăÎÂʱĽä¶ÔÝÍȡÂʵÄÓ°Ď첻´óˇŁĽÓČČ20ˇ«50 minŁ¬ÝÍȡÂĘľůżÉ´ď100%Ł¬ĽĚĐřĽÓČČŁ¬ÝÍȡÂĘÓÖ»áĎ½µŁ¬ŐâżÉÄÜĘÇŇňÎŞĽĚĐřĽÓČČ×č°ÁËÜĹ˝řČëSDS˝şĘřĎࡣËć×ĹşăÎÂʱĽäŃÓł¤Ł¬ÄýľŰĎŕÖđ˝ĄĹ¨ËőŁ¬Ďŕ±ČÖ𽥼őСŁ¬ÔÚşăÎÂ50
minʱŁ¬şă¶¨˛»±äˇŁąĘʵŃéѡÔńşăÎÂʱĽäÎŞ50
minˇŁ
2.2.4 ×ǵăÝÍȡµÄ×îÓĹĚőĽţ
×ŰÉĎ˝áąűŁ¬×ǵăÝÍȡµÄ×îÓĹĚőĽţÎŞŁş2ŁĄSDSŁ»HClŨ¶Č4.2
mol·L-1Ł»NaClŨ¶Č0.31 mol·L-1Ł»Ć˝şâζČ50
ˇćŁ»Ć˝şâʱĽä50 minˇŁ
2.3 ÜĹ-Ľ×´ĽČÜŇşµÄ±ę׼ą¤×÷ÇúĎß
׼ȷĹäÖĆ7¸öŨ¶ČĚݶȵÄÜĹ-Ľ×´Ľ±ę׼ҺŁ¬˝řĐĐÉ«Ć×·ÖÎöŁ¬¸ůľÝ·ĺĂć»ý¶ÔĆ京Áż»ćÖƱę׼ÇúĎߡŁĎßĐԻع鷽łĚÎŞy=438.98533x-8.75855Ł¬ĎŕąŘϵĘýR2=0.9994Ł¬ĎßĐÔ·¶Î§0ˇ«10.0 mg·L-1ˇŁ
2.4·˝·¨Ľě˛âĎ޵IJⶨ
·˝·¨Ľě˛âĎŢ(method detection limitŁ¬MDL)µÄČ·Á˘˛ÎŐŐÎÄĎ×[20]Ł¬Ľ´»ůµ×ĽÓ±ęĘÔŃůľŔúÔ¤´¦ŔíÓëŇÇĆ÷·ÖÎöČ«ąýłĚŁ¬ľn(ˇÝ4)´ÎĆ˝ĐвⶨŁ¬ĽĆËăĆä±ę׼ƫ˛îs
Ł¬ÔňMDL=s tf,0.01(µĄ±ß)Ł˝s tf,0.02(Ë«±ß)ˇŁĽĆËăµĂµ˝µÄMDLÓëĽÓ±ęŨ¶Č˝řĐбȽϣ¬ČçąűĽÓ±ęŨ¶Č£
5MDLŁ¬ÔňŇÔÉĎąýłĚÓĐЧŁ¬·ńÔňŁ¬µ÷ŐűĽÓ±ęŨ¶ČŁ¬ÖŘĐ²ⶨÓëĽĆËăMDLˇŁ
ŇÔMilli-QˮΪŃůĆ·»ůµ×Ł¬ÜĹ-Ľ×´ĽĹ¨¶ČÎŞ0.05 mg·L-1Ł¬Ć˝Đвⶨ4´ÎŁ¬Ć˝ľůֵΪ0.070
mg·L-1Ł¬SDÎŞ0.003 mg·L-1Ł¬˛é±ít3,
0.01(µĄ±ß)ÎŞ4.541Ł¬ĽĆËăµĂ±ľ·˝·¨µÄĽě˛âĎŢMDL = S.D. ˇÁt3, 0.01(µĄ±ß)=0.0136
mg·L-1Ł¬ĽÓ±ęŨ¶Č(0.05 mg·L-1)СÓÚ5±¶µÄMDLÖµ(0.068 mg·L-1)Ł¬ĽĆËăËůµĂµÄ·˝·¨Ľě˛âĎŢÓĐЧˇŁ
2.5 »ůµ×ĽÓ±ęʵŃ鼰ŃůĆ·˛â¶¨
ÎŞŃéÖ¤¸Ă·¨µÄżÉĐĐĐÔŁ¬ÔÚ×îĽŃĚőĽţĎÂŁ¬ŃˇÔńMilli-QË®ˇ˘×ÔŔ´Ë®şÍşţË®ŃůĆ·˝řĐвⶨĽ°»ůµ×ĽÓ±ęʵŃ飬żĽ˛ěÁË·˝·¨µÄ»ŘĘŐÂʺ;«Ăܶȣ¬˝áąűĽű±í1ˇŁ
±í1 ʵĽĘŃůĆ·şÍĽÓ±ę»ŘĘŐÂĘʵŃé˝áąű
(nŁ˝3)
Tab. 1 Results of real sample and recoveries
of spiked samples (n=3)
Ë®Ńů |
ĽÓČëŨ¶Č (mg·L-1) |
Ć˝ľůÖµˇŔ SD |
ĽÓ±ę»ŘĘŐÂĘ (%) |
RSD(%) |
Milli-Q Ë® |
0 |
ND |
Ł |
Ł |
| ˇˇ | 0.2 |
0.188ˇŔ0.003 |
94.0 |
1.60 |
| ˇˇ | 1.0 |
1.051ˇŔ0.005 |
105.1 |
0.48 |
| ˇˇ | 3.0 |
2.820ˇŔ0.232 |
94.0 |
8.23 |
ÇďÖĐşţË® |
0 |
ND |
Ł |
Ł |
| ˇˇ | 0.2 |
0.198ˇŔ0.003 |
99.0 |
1.52 |
| ˇˇ | 1.0 |
0.962ˇŔ0.002 |
96.2 |
0.21 |
| ˇˇ | 3.0 |
2.734ˇŔ0.077 |
91.1 |
2.82 |
¶«şţË® |
0 |
ND |
Ł |
Ł |
| ˇˇ | 0.2 |
0.193ˇŔ0.003 |
96.5 |
1.56 |
| ˇˇ | 1.0 |
1.024ˇŔ0.006 |
102.4 |
0.59 |
| ˇˇ | 3.0 |
2.604ˇŔ0.168 |
86.8 |
6.45 |
×ÔŔ´Ë® |
0 |
ND |
Ł |
Ł |
| ˇˇ | 0.2 |
0.180ˇŔ0.002 |
90.0 |
1.11 |
| ˇˇ | 1.0 |
0.904ˇŔ0.017 |
90.4 |
1.89 |
| ˇˇ | 3.0 |
2.422ˇŔ0.143 |
80.7 |
5.91 |
עŁş
NDΪδĽěłöˇŁ3 ˝áĘřÓď
REFERENCES
[1] şú˝ˇ.ąóŃôĘĐ´óĆř-Ë®Ěĺ-ÍÁČŔ»·ľłÖж໷·ĽĚţµÄŃĐľż[D].ÖĐąúżĆѧԺµŘÇň»ŻŃ§ŃĐľżËů,2005.
[2] Paleologos E K, Giokas D L, Karayannis M I. Trends in Analytical Chemistry, 2005, 24(5): 426-436.
[3] Li J L, Chen B H. Journal of Colloid and Interface Science, 2003, 263: 625-632.
[4] Giokas D L, Eksperiandova L P, Blank A B et al. Analytica Chimica Acta, 2004, 505: 51-58.
[5] Sirimanne S R, Barr J R, Patterson D G et al. Anal. Chem, 1996, 68: 1556-1560.
[6] Fang Q, Yeung H W, Leung H W et al. Chromatogr A, 2000, 904: 47-55.
[7] Garcia-Pinto C, Perez-Pavon J L, Moreno-Cordero B.Anal. Chem, 1995, 67: 2606-2612.
[8] Wu Y C, Huang S D. Anal. Chim. Acta, 1998, 373: 197-206.
[9] Casero I, Sicilia D, Rubio S et al. Anal. Chem, 1999(71): 4519-4526.
[10] Saitoh T, Hinze W L. Anal. Chem, 1991, 63: 2520-2525.
[11] Eiguren-Fernandez A, Sosa-Ferrera Z, Santana-Rodriguez J J. Anal. Chim. Acta, 1998, 358: 145-155.
[12] Sirimanne S R, Patterson D G, Ma L et al. Chromatogr, B 1998, 716: 129-137.
[13] Eiguren-Fernandez A, Sosa-Ferrera Z, Santana-Rodriguez J J. Analyst, 1999, 124: 487-494.
[14] Ba'rbara D, Veroˇänica P, Juan H. Analyst, 2005, 130: 571-577.
[15] ÂíÔŔ,»ĆżĄĐŰ.ÉĎşŁ»·ľłżĆѧ, 2000, 19(7): 319-324.
[16] Francisco M, Soledad R, Dolores P B. Journal of Chromatography A, 2002, 962: 1-8.
[17] Sicilia D, Rubio S, Pe?rez-Bendito D et al. Analytica Chimica Acta, 1999 , 392: 29-38.
[18] Theodosios I, Sikalos, Evangelos K. P. Anal. Chem, 2005, 77: 2544-2549.
[19] Nascentes C C,Arruda M A Z. Talanta, 2003, 61: 759-768.
[20] Berger W, McCarty H, Smith R K. Environmental Laboratory Data Evaluation[M].GA,the Unites States:Genium Publishing Corporation, 1996: 2-11.
ˇˇ