http://www.chemistrymag.org/cji/2008/107033pc.htm |
Jul.
1, 2008 Vol.10 No.7 P.33 Copyright |
(Department of Chemistry, Jinan University, Guangzhou, 510632)
Abstract Ti-SBA-15 photocatalysts have been synthesized by a direct-synthesis step under hydrothermal conditions using titanium butoxide and titanium as titanium precursor, respectively. The results showed that the hydrolysis rate of Ti(SO4)2 is much higher than TEOS in our experimental conditions, leading to the formation of TiO2 phase out of the SBA-15 crystalline network. The addition of chelating agents acetylacetone forms complexes with the Ti ions and slow down the hydrolysis of the highly reactive titanium butoxide. However the products followed by 600ºC
heat treatment were obtained with both anatase and rutile phase. The Ti-SBA-15-1 catalysts with Si/Ti weight ratio of 1.3 show highest photocatalytic activity and under 60 min UV light irradiation, 93.7% of methyl blue in an aqueous dispersion was photodegraded.Keyword hydrothermal method, photocatalysts, direct-synthesis 水热法一步法合成Ti-SBA-15光催化剂的研究
钟慧琼,李明,张丽珍,杨骏
(暨南大学化学系,广州,510632)
摘要
分别以硫酸钛和钛酸丁酯为钛源,正硅酸乙酯为硅源,采用水热法一步合成了Ti-SBA-15光催化剂。结果表明:在强酸性条件下硫酸钛的水解速度较正硅酸乙酯快,导致了部分的TiO2堆积在硅基质的外部;而用乙酰丙酮作为钛酸正丁酯水解抑制剂然能使得两者的水解速度相当,但是最后600
℃热处理得到的样品都是金红石和锐钛矿相的混晶。Si:Ti=1.3 的Ti-SBA-15-1样品有较高的光催化活性,60
min紫外光照下亚甲基蓝的脱色率为93.7%。
关键词 水热法 光催化剂
随着染料工业的迅速发展, 其带来的污染问题也日益严重,全世界生产的染料约有15%在生产或应用过程中被排放到废水中[1]。染料废水处理方法有化学、物理和生物法等,其中光催化氧化是近年来研究的热点。二氧化钛以其价廉易得、容易处理、高催化活性、化学性质稳定和耐光腐蚀性的特点使它成为了光催化剂的首选[2]。但单独使用二氧化钛时,由于一般二氧化钛的颗粒较小,使得其回收困难,从而限制了它的工业应用[3]。鉴于此,有文献报道把二氧化钛负载到各种基质上,如SiO2,利用其比表面和孔容大、紫外吸收少、吸附容量大等特性以提高催化剂对有机污染物的矿化能力,减少中间产物的浓度[4]。因此,TiO2-SiO2复合氧化物成为了非常有潜力的光催化剂。最近,我们采用两步法成功制备了光催化降解染料性能优越的TiO2-SiO2催化剂[5-6]。众所周知,制备方法很大程度决定了二氧化钛的晶相组成、晶粒大小和形貌等,从而影响了二氧化钛的光催化性能。因此,本文拟分别以硫酸钛和钛酸丁酯为钛源,正硅酸乙酯为硅源,采用水热一步合成Ti-SBA-15光催化剂,系统研究制备条件对TiO2-SiO2催化剂可见光光催化性能的影响。
2
实验部分
2.1 Ti-SBA-15催化剂的制备
硫酸钛为钛源:将2 g P-123溶于60 ml HNO3(2
mol/L)溶液,同时加入定量的硫酸钛以及3.7 g正硅酸乙酯,在40
℃下搅拌1 h,然后转入聚四氟乙烯水热罐中,在100 ℃下水热72 h。取出产物,离心分离,干燥,最后得到的产物在600
℃下热处理5 h,所得样品记为Ti-SBA-15-1。催化剂中TiO2的含量通过H2O2显色分光光度法测定
钛酸丁酯为钛源:将2 g P-123溶于60 ml HCl(2 mol/L)溶液,再同时加入定量的异丙醇钛和乙酰丙酮混合液(摩尔比,钛酸丁酯:乙酰丙酮=1: 0.6)以及2.08 g正硅酸乙酯,在40 ℃下搅拌0.5 h,出现部分沉淀。继续搅拌24 h,然后转入聚四氟乙烯水热罐中,在100 ℃下水热24 h。取出产物,离心分离,干燥,最后得到的产物在600 ℃下热处理5 h,所得样品记为Ti-SBA-15-2。
2.2 催化剂表征
XRD采用MSAL XD-2 型X射线衍射仪(布莱格科技(北京)有限公司)测定分析样品的物相,辐射源为:CuKa,0.15418 nm,电压36KV,电流24 mA,扫描速度:4°/min,步宽为0.02°,扫描范围2q=20-60°。
2.3 光催化实验
100 ml浓度为20 mg/l的亚甲基蓝为降解对象,首先加入0.05 g的催化剂在室温下暗室搅拌15 min,以达到吸附平衡。再放置于紫外灯下进行降解,循环水浴控制反应温度,同时以90 ml/min的速度向溶液中鼓入空气。反应每隔15 min 取样2 ml,离心分离后用紫外-可见分光光度仪测定亚甲基蓝的剩余浓度。
3.1 X射线衍射分析 (XRD)
图1 和2分别为不同硅钛比的Ti-SBA-15-1 和Ti-SBA-15-2的XRD谱图,从图可见,600 ℃热处理的Ti-SBA-15-1样品都仅出现锐钛矿相,且随着二氧化钛含量的增加,锐钛矿的特征吸收峰变的更加尖锐。 600 ℃热处理的Ti-SBA-15-2样品都呈现出金红石和锐钛矿相的混晶相,与Ti-SBA-15-1有显著的差异。根据Scherrer 公式计算出的TiO2平均粒径结果表明,对于Ti-SBA-15-2的样品,随着钛含量的增加,二氧化钛的晶粒有所变大,但是变化不明显,相对于Ti-SBA-15-1样品来说,其晶粒要小得多。同是Si:Ti=1的催化剂,两种方法合成的样品晶粒大小相差近5倍,可能的原因是硫酸钛相对于正硅酸乙酯在强酸性条件下水解速度要快一些,而添加了乙酰丙酮的钛酸丁酯的水解速度却和正硅酸乙酯相匹配[8]。硫酸钛的快速水解导致了二氧化钛颗粒在二氧化硅孔道外的大量堆积,分散不均匀,导致了热处理过程中二氧化钛粒子的团聚,因而晶粒更大。
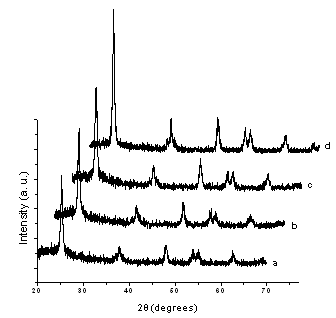
图1 Ti-SBA-15-1样品的XRD谱图,硅钛质量比为:
(a)4;(b)2;(c)1.3;(d)1
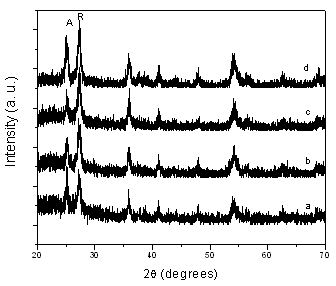
图2 Ti-SBA-15-2样品的XRD谱图,硅钛质量比为:
(a)3;(b)1.5;(c)1;(d)0.5
Gopal等
[9]认为二氧化钛晶相的形成主要取决于TiO2的成核和生长过程。如果聚合发生在水解完全以前,可能形成无定形的或是亚稳定状态的锐钛矿二氧化钛[10]。在酸性条件下,水解速度得到提高[11],相反聚合的速度降低了,另外依据金红石和锐钛矿在结构上的差异,在强酸性条件下也有利于金红石二氧化钛的形成[12],在低酸度下有利于锐钛矿的形成。在前期的工作中发现二氧化硅对二氧化钛热处理的过程中起到了抑制金红石相生成的作用[5-6]。但本文用添加了乙酰丙酮的钛酸丁酯作为钛源的反应却得到了金红石相的二氧化钛,可能原因是:乙酰丙酮作为水解抑制剂,极大降低了钛酸丁酯的水解速率,在成核的过程中出现更加稳定的金红石相。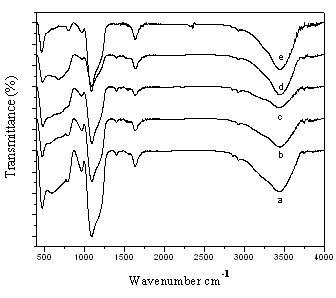
图3 Ti-SBA-15-1样品FT-IR谱图,硅钛质量比为:
(a)4;(b)3;(c)1.3;(d)1;(e)纯SBA-15
3.2 FT-IR光谱分析
图3和4分别是不同硅钛比的Ti-SBA-15-1和 Ti-SBA-15-2的FT-IR谱图。谱中1104 cm-1处的吸收峰是由Si-O-Si键的不对称伸缩振动引起的;与此相对在800和470 cm-1处的吸收峰分别是由Si-O-Si键的对称伸缩振动和弯曲振动引起的[13]。450-480 cm-1 是二氧化钛中Ti-O-Ti的振动,一般认为930-970 cm-1被认为是Ti-O-Si振动,而Ti-O-Si 是判断二氧化钛是否与二氧化硅有键结合的主要依据[14] 。由图可见,所有样品的谱图在930-970 cm-1内都出现了明显的吸收峰,可以证明Ti-O-Si的形成,随着Ti含量的增加这部分的吸收峰又开始慢慢变小,最后已经基本消失(图3)。
对于Ti-SBA-15-1样品,随着Ti含量的增加,700-860 cm-1慢慢出现吸收峰,这归咎于没有进入孔道而分散聚集在外的TiO2所形成的特征峰[15]。这和XRD结果的分析一致,硫酸钛的快速水解使得部分Ti并没有进入分子筛的骨架中,而是相互堆积成TiO2颗粒。
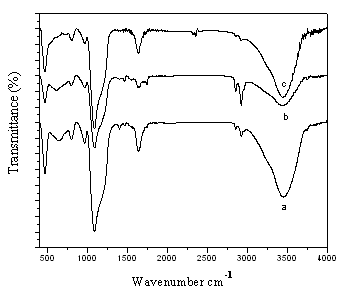
图4 Ti-SBA-15-2样品FT-IR谱图,硅钛质量为:
(a)3;(b)1.5;(c)纯SBA-15 图5 不同硅钛比的Ti-SBA-15-1 和P-25的光催化降解曲线
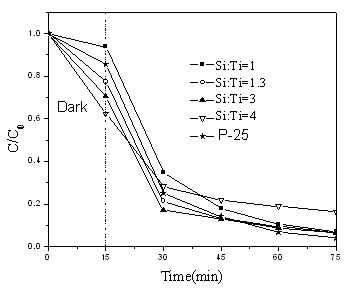
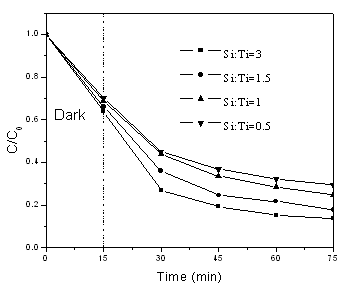
图6 不同硅钛比的Ti-SBA-15-2光催化降解曲线 图5和6表示亚甲基蓝的脱色率随时间的变化关系图。由图可知,当Si:Ti=1.3 时的Ti-SBA-15-1样品有着和P-25相近的光催化活性,60 min紫外光照下亚甲基蓝的脱色率为93.7%。不同的硅钛比的样品在最初的15 min 的暗室吸附条件下,随着Ti含量的增加,比表面下降,吸附能力也降低。众所周知,除了吸附能力外,晶型、晶粒大小以及结晶度也是影响光催化能力的重要因素。因此当硅钛比达到最佳时,催化剂体现出最好的光催化活性,许多研究结果也证实这一点[5,16]。而对于Ti-SBA-15-2样品,几种硅钛比的样品的光催化活性都不高,并且随着硅钛比的升高有降低的趋势。Ti-SBA-15-2样品光催化活性不高可能是因为金红石相的出现,很多研究都指出对于大多数反应来说锐钛矿表现出比金红石型TiO2 更高的光催化活性光催化活性,这可能是由于锐钛矿具有较高的费米能级,低的氧容量以及高程度的羟基化作用[17] 。
通常染料在TiO2光催化剂上的降解符合一级反应,本文将暗吸附15 min达到吸附平衡后的亚甲基蓝浓度作为初始浓度,各催化剂的亚甲基蓝降解反应速率常数列于表1。从表可见,无论是用硫酸钛作为钛源还是用钛酸正丁酯为钛源,一步合成法得到的Ti-SBA-15都没有表现出优异的光催化活性,低于P-25。对于硫酸钛作钛源来说,其水解速率在本实验条件下较正硅酸乙酯更快,导致了部分的TiO2堆积在硅基质的外部,其光催化活性相对较高;而用乙酰丙酮作为水解抑制剂来限制钛酸正丁酯虽然能使得两者的水解速度相当,但是最后得到的都是金红石和锐钛矿相的混晶,且结晶度较差,导致低的光催化活性。在我们之前两步法的工作中[5], 由于钛源是加入在已经预先成型的硅胶中,相对一步法而言,这时水解后的钛物种很难均匀地进入无机硅-有机复合物种之间,形成无机硅 + 钛有机复合物种,因此钛在介孔墙壁中的分散相对一步法而言是不均匀的,聚集态的钛比较多,而这可能正是其光催化性能优越的原因之一。
表1 各催化剂对亚甲基蓝的降解速率常数
Ti-SBA-15-1 |
速率常数/min-1 |
Ti-SBA-15-2 |
速率常数/min-1 |
Si:Ti=4 |
0.026 |
Si:Ti=3 |
0.030 |
Si:Ti=3 |
0.046 |
Si:Ti=1.5 |
0.025 |
Si:Ti=1.3 |
0.047 |
Si:Ti=1 |
0.019 |
Si:Ti=1 |
0.047 |
Si:Ti=0.5 |
0.017 |
4 结论
分别以硫酸钛和钛酸丁酯为钛源,正硅酸乙酯为硅源,采用水热一步合成了Ti-SBA-15光催化剂,结果发现两种方法合成的含Ti分子筛并没有表现出很好的光催化活性。解决问题的关键在于如何找到合适的方法使得钛源和硅源两者的水解速度相匹配;此外一步合成法得的Ti较为均匀的分散在整个复合氧化物中,但是光催化反应的活性位点却出现在材料的表面,因此有理由相信负载型的硅钛复合氧化物可能会表现出更好的光催化活性。
[1] Houas A, Lachheb H, Ksibi M et al. Appl. Catal. B: Environ. 2001,31: 145
[2] Konstantinou I. K, Albanis T. A. Appl. Catal. B: Environ. 2004,49: 1
[3] Pizarro P, Guillard C, Perol N et al. Catal Today 2005, 101: 211
[4] Tokimoto T., Ito S., Kuwabata S. et al. Environ. Sci. Technol.1996, 30: 1275
[5] Yang J, Zhang J, Zhu L W et al. J. Hazardous Mater. 2006, 137( 2): 952-958
[6] Yang J, Zhu L W, Zhang J, Zhu L W et al. React. Kinet. Catal. Lett. 2007, 91: 21-28
[7] 胡春,王怡中,汤鸿霄. 催化学报,2001,22:185-188
[8] 朱金华, 沈伟, 徐华龙等.化学学报,2003, 61(2), 202-207
[9] Gopal M, Mobey W J, Chan L C et al. J. Mater. Sci. 1997, 32: 6001.
[10] Yoon K H, Noh J S, Kwon C H et al. Mater. Chem. Phys. 2006, 95 (1): 79-83
[11] Brinker C J. J. Non-Cryst. Solids 1988, 100 : 31.
[12] Livage I, Henry M, Sanchez C. Solid State Chem. 1988, 18, 259.
[13] Jung K Y, Park S B. Appl. Catal. B 2000, 25, 249.
[14] Lee D W, Ihm S K, Lee K H. Chem. Mater. 2005, 17 :4461
[15] Armaroli T, Milella F, Notari B et al. Topic. Catal. 2001, 15, 63
[16] Li Z J, Hou B, Xu Y et al. J. solid state chem. 2005, 178 :1395.
[17] Linsbigler A L, Lu G Q, Yates Jr. J T. Chem. Rev. 1995, 95:735.