http://www.chemistrymag.org/cji/2008/109044pe.htm |
Sep.
1, 2008 Vol.10 No.9 P.44 Copyright |
Zhang
Siyong, Zhao Qingfei
(Department of Chemistry, Shanghai Normal University, Shanghai 200234, China)
Abstract (S)-(-)-2-(4-chlorophenyl)-3-methylbutyric acid was separated from its
antipode using (S)-(-)-a–methylbenzylamine as
chiral separation, series of new compounds
(S)-(-)-5,7-disubstitute-2-(4-chlorophenyl-3-methylbutanimine)-2H-1,2,4-thiadiazolo[2,3-a]pyrimidine
derivatives were obtained using TBAB (Tetrabutyl ammonium bromide) as solid-liquid phase
transfer catalyst (PTC) under water and hydrogen peroxide as oxidant. Their structures
were exactly confirmed by IR, 1H NMR and elemental analysis. Our method using
hydrogen peroxide as oxidant under the condition of PTC in this paper has the advantage of
short reaction time, good reaction yield and environmentally friendly process compared to
the classical method.
Keyword Clean synthesis, TBAB, Pyrimidine derivatives
1 INTRODUCTION
2H-1, 2, 4-thiadiazolo [2, 3-a]
pyrimidine derivatives are a novel group of synthetic herbicidal agents [1-3],
which are active against the weeds of Digitaria sanguinalis (L) Scop, and Chenopodium.
In general, these compounds behave in a manner similar to that of sulfonylurea herbicides,
inhibiting the synthesis of acetolactate, which has become a very attractive target for
herbicides [4-7]. Moreover, because of 2H-1, 2, 4-thiadiazolo [2, 3-a]
pyrimidine derivatives containing an inherently weak N-S bond which benefits plant’s absorption and metabolism, it’s selective activities are
superior to the sulfonylurea [8]. (S)-(-)-2-(4-chlorophenyl)-3-methylbutyric
acid compounds have also attracted the attention of many investigators due to
insecticidal, antibacterial, pesticidal activities and promoting effect on plant growth [9-11].In
recent years, organic reactions in water have been the object of growing efforts since
water is cheap, safe and a clean solvent [12-13]. In fact, organic solvent
removal is of paramount importance to minimize economic cost and environmental impact of
chemical processes. A survey of literature shows that many organic reactions have recently
been accelerated by PTC and that hydrogen peroxide, which is the most environmentally, and
economically attractive oxidant, in place of bromine, is to generate the cyclization in
water.
The title compounds were synthesized by the method in Scheme 1.
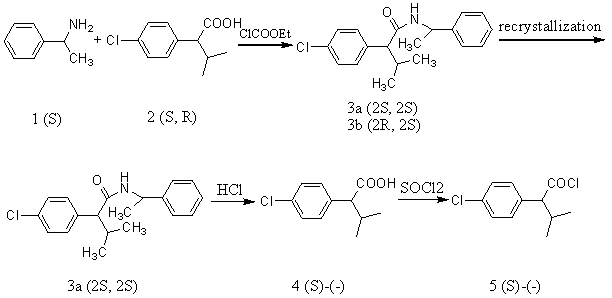
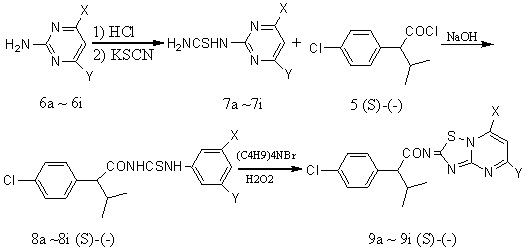
Table 1 The Substituents of Target Compounds 9a-9i
Compd |
a |
b |
c |
d |
e |
f |
g |
h |
i |
X |
CH3 |
OCH3 |
Cl |
Cl |
OH |
Cl |
Cl |
OC2H5 |
CH3 |
Y |
CH3 |
OCH3 |
Cl |
OCH3 |
CH3 |
OC2H5 |
CH3 |
OC2H5 |
OCH3 |
2 RESULTS AND DISCUSSION
(S)-(-)-2-(4-chlorophenyl)-3-methylbutyric acid was separated from its antipode using
(S)-(-)-a–methyl benzyl amine as chiral separation.
The intermediate (S)-(-)N-[3-methyl-2-(4-chlorophenyl)butyryl]-N'-(2-pyrimidinyl)
thioureas (8) were synthesized by the reaction of
(S)-(-)2-(4-chlorophenyl)-3-methylbutyl acyl chloride and
4,6-disubstituted-pyrimidine-2-yl-thiourea. Series of compounds (S)-(-) (9) were
obtained by oxidizing cyclization of corresponding compounds (S)-(-) (8), using
TBAB as PTC catalyst and hydrogen peroxide as oxidant in water.
4, 6-Disubstituted-2-amino-pyrimidine was partially soluble in water.
Using HCl as acid (6 molar equiv.) allowed the 4, 6-disubstitute-2-amino-pyrimidine to
sufficiently soluble in water. Potassium thiocyanide was added to the reaction, the
mixture stirred at 90 - 100 ℃ for several hours, (7)
was obtained after the reaction mixture was left to cool. (8) were synthesized
after acyl chloride was added to a solution of the (7) in 10 % NaOH solution and
prolonged for 1 ~ 2 h.. In some reports, (9) have been prepared oxidizing
cyclization (8), bromine as oxidant in chloroform.. In order to achieve an even
more efficient and environmentally friendly process, we planned to develop an advanced
procedure enabling the synthesis of 5, 7- disubstitute-2H-1, 2, 4-thiadiazolo [2, 3-a]
pyrimidine by oxidizing cyclization without bromine, possibly without use of any organic
solvent throughout the whole process .Oxidizing cyclization of (8) in water at 0 -
2 ℃ with H2O2 (1.5 - 2.0 molar
equiv.) and the mixture continued to stirred at ambient for 1 ~ 2 h to ensure complete
conversion to (9) , as the result of this, 40 ~ 60 % only of Oxidizing cyclization
(9) was obtain. By using of PTC catalyst to the procedure; the yield of (9)
was further improved. The best result of the process as obtained by using 1.5 % TBAF
(Tetrabutyl ammonium fluoride) only, to generate 90 - 95 % overall yield of (9).
Although good yield of Oxidizing cyclization was obtained, TBAF is a quite expensive
compound. In order to develop a practical and economical procedure, using of TBAB in place
of TBAF, 75 - 80 % of (9) was isolated. In summary, oxidant of hydrogen peroxide in
place of bromine and the use of catalytic amounts of TBAB, generated products of 5,7-
disubstituted-acyl-2H-1, 2, 4-thiadiazolo[2,3-a]pyrimidine. The new method is
atomically efficient and environmentally friendly since no organic solvent is required
through the whole procedure and pure products can be isolated.
All the structures of the newly synthesized compounds (9) were
elucidated and confirmed by elemental analyses (Table 2), 1H NMR (Table
3) and optical rotatios data (Table 4). The IR (KBr) spectrum displayed
absorptions at about 1690 cm-1, 1600 cm-1 and 1370 cm-1,
which are assigned for C=O, C=N and CH(CH3)2 functions
respectively. The 1H NMR (DMSO-d6) spectrum revealed signals at
about δ 0.80 (d, 3H, CH(CH3)2),
Table 2. Physical Constants of Compounds 9a-9i
Compd. |
Molecular |
Appearance |
M.P. |
Yield (%) |
Analytical data |
||
Formula |
C |
H |
N |
||||
9a |
C18H19ClN4OS |
white powder |
254 ~ 256 |
78 |
57.60 |
5.00 |
14.87 |
374.5 |
(57.68) |
(5.07) |
(14.95) |
||||
9b |
C18H19ClN4O3S |
white powder |
252 ~ 255 |
80 |
53.10 |
4.60 |
13.70 |
406.5 |
(53.13) |
(4.67) |
(13.77) |
||||
9c |
C16H13Cl3N4OS |
white powder |
245 ~ 257 |
76 |
46.11 |
3.10 |
13.40 |
415.5 |
(46.21) |
(3.13) |
(13.47) |
||||
9d |
C17H16Cl2N4O2S |
white powder |
251 ~ 253 |
75 |
49.56 |
3.84 |
13.70 |
411 |
(49.63) |
(3.89) |
(13.76) |
||||
9e |
C17H17ClN4O2S |
white powder |
>260 |
80 |
54.05 |
4.40 |
14.80 |
376.5 |
(54.18) |
(4.51) |
(14.87) |
||||
9f |
C18H18Cl2N4O2S |
white powder |
231 ~ 233 |
79 |
50.70 |
4.16 |
13.10 |
425 |
(50.82) |
(4.23) |
(13.17) |
||||
9g |
|
white powder |
235 ~ 237 |
80 |
51.54 |
3.95 |
14.10 |
395 |
(51.64) |
(4.05) |
(14.18) |
||||
9h |
C20H23ClN4O3S |
light yellow powder |
251 ~ 253 |
77 |
55.60 |
5.18 |
12.80 |
434.5 |
(55.23) |
(5.29) |
(12.89) |
||||
9i |
C18H19ClN4O2S |
light yellow powder |
211 ~ 214 |
79 |
55.20 |
4.76 |
14.30 |
390.5 |
(55.31) |
(4.86) |
(14.34) |
||||
Table 3. 1H NMR data of Compounds 9a-9i *
Compd |
1 H NMR (d, DMSO ) |
9a |
0.70 (d, J= 7 Hz, 3H, CH(CH3)2), 0.90 (d, J= 8 Hz, 3H, CH(CH3)2), 2.20 ~ 2.40 (m, 1H, CH(CH3)2), 3.90 ~ 4.10 (m, 1H, CHCH(CH3)2), 7.30 ~ 7.50 (m, 4H, Ph-H), 2.10 (s, 3H, CH3 ), 2.20 (s, 3H, CH3), 5.94 (s, 1H, py-5'-H) |
9b |
0.80 (d, J= 7 Hz, 3H, CH(CH3)2), 1.00 (d, J= 8 Hz, 3H, CH(CH3)2), 2.10 ~ 2.30 (m, 1H, CH(CH3)2), 3.80 ~ 4.00 (m, 1H, CHCH(CH3)2), 7.20 ~ 7.40 (m, 4H, Ph-H), 4.41 (s, 3H, OCH3 ), 4.60 (s, 3H, OCH3), 5.91 (s, 1H, py-5'-H) |
9c |
1.0 (d, J= 7 Hz, 3H, CH(CH3)2),1.20 (d, J= 8 Hz, 3H, CH(CH3)2), 2.45 ~ 2.55 (m, 1H, CH(CH3)2), 3.90 ~ 4.00 (m, 1H, CHCH(CH3)2), 7.30 ~ 7.40 (m, 4H, Ph-H), 5.82 (s, 1H, py-5'-H) |
9d |
1.00 (d, J= 7 Hz, 3H, CH(CH3)2), 1.10 (d, J= 8 Hz, 3H, CH(CH3)2 ), 2.30 ~ 2.40 (m, 1H, CH(CH3)2), 3.90 ~ 4.00 (m, 1H, CHCH(CH3)2), 7.50 ~ 7.60 (m, 4H, Ph-H), 4.15 (s, 3H, OCH3 ), 5.74 (s, 1H, py-5'-H) |
9e |
0.90 (d, J= 7 Hz, 3H, CH(CH3)2), 1.10 (d, J= 8 Hz,3H, CH(CH3)2), 2.40 ~ 2.55 (m, 1H, CH(CH 3)2), 3.90 ~ 4.05 (m, 1H, CHCH(CH3)2), 7.30 ~ 7.50 (m, 4H, Ph-H), 2.49 (s, 3H, CH3), 6.87 (s, 1H, py-5'-H) |
9f |
1.00 (d, J= 7 Hz, 3H, CH(CH3 )2), 1.10 (d, J= 8 Hz, 3H, CH(CH3)2), 2.30 ~ 2.40 (m, 1H, CH(CH3)2), 3.90 ~ 4.00 (m, 1H, CHCH(CH3)2), 7.50 ~ 7.60 (m, 4H, Ph-H), 4.29 ~ 4.40 (q, 2H, OCH2CH3 ), 1.68 ~ 1.80 (t, 3H, OCH2CH3 ), 5.74 (s, 1H, py-5'-H) |
9g |
1.00 (d, J= 7 Hz, 3H, CH(CH 3)2), 1.10 (d, J= 8 Hz, 3H, CH(CH3)2), 2.30 ~ 2.40 (m, 1H, CH(CH3)2), 3.90 ~ 4.00 (m, 1H, CHCH(CH3)2), 7.50 ~ 7.60 (m, 4H, Ph-H), 2.11 (s, 3H, CH3 ), 5.74 (s, 1H, py-5'-H) |
9h |
0.90 (d, J= 7 Hz, 3H, CH(CH3)2), 1.10 (d, J= 8 Hz, 3H, CH(CH3)2), 2.40 ~ 2.55 (m, 1H, CH(CH3)2 ), 3.90 ~ 4.05 (m, 1H, CHCH( CH3)2 ), 7.30 ~ 7.50 (m, 4H, Ph-H), 2.40 (s, 3H, CH3 ), 4.29 ~ 4.40 (q, 2H, OCH2CH3), 1.68 ~ 1.80 (t, 3H, OCH2CH3), 6.87 (s, 1H, py-5'-H) |
9i |
1.00 (d, J= 7 Hz, 3H, CH(CH3)2), 1.10 (d, J= 8 Hz, 3H, CH(CH3)2), 2.30 ~ 2.40 (m, 1H, CH(CH3)2), 3.90 ~ 4.00 (m, 1H, CHCH(CH3)2), 7.50 ~ 7.60 (m, 4H, Ph-H), 1.80 (s, 3H, CH3 ),4.00 (s, 3H, OCH3), 5.74 (s, 1H, py-5'-H) |
*Abbreviations: s = singlet, t = triplet, q = quarterlet, m = multiplet, Py = pyrimidine
Table 4 Optical rotatios data of Compounds 9a-9i
Compd |
Temperature(℃) |
L (dm) |
C (mg/100ml) |
[a]20D |
solvent |
9a |
20 |
0.2 |
1.15 |
-10.2 |
methanol |
9b |
20 |
0.2 |
0.90 |
-19.5 |
methanol |
9c |
18 |
0.2 |
1.00 |
- 19.8 |
methanol |
9d |
23 |
0.2 |
1.80 |
-10.4 |
methanol |
9e |
20 |
0.2 |
0.50 |
-12.5 |
methanol |
9f |
20 |
0.2 |
1.24 |
-9.5 |
methanol |
9g |
25 |
0.2 |
0.80 |
-14.9 |
methanol |
9h |
20 |
0.2 |
1.20 |
-20.8 |
methanol |
9i |
20 |
0.2 |
0.70 |
-15.5 |
methanol |
3 EXPERIMENTAL SECTION
All starting materials are commercial products of chemical or analytic grade purity.
Sulfuric chloride was distillated before use and potassium thiocyanate was baked before
use. 4,6-disubstitute-2-amino-pyrimidine (6) was prepared by the literature method. 8a~8i
were obtained by the literature method. The melting points were determined on an XT4A
micro digital melting point apparatus and are uncorrected. The isolated compounds (9) were
characterized by elemental microanalyses. The C, H and N analyses were repeated twice. The
result of elemental analyses is listed in Table 2. IR spectra were obtained on a
Nicolet5DX FT-IR spectrophotometer in the region 4000-400cm-1 KBr discs. 1H
NMR spectra were recorded on a Bruker Av 400 MHz spectrometer with CDCl3 or d6-DMSO
as the solvent. Chemical shift values are reported in ppm (
3.1 Preparation and separation of the diastereomer N- (1-phenylethyl)-2-(4-chlorophenyl) -3-methylbutyramide(3)
To a solution of (±) 2-(4-chlorophenyl)-3-methylbutric acid (42.5 g, 0.2 mol) in THF(300 ml)was added dropwise triethylamine (21.3 g, 0.21 mol) and ClCO2Et (22.8 g, 0.21 mol) at 0 ℃ over a period of 20 min. Then (S)-(-)-a-methylbenzylamine (25.4 g, 0.21 mol) was added and the reaction was stirred for 3 h at 0 ℃. After filtration, concentration and recrystallization from MeOH/H2O (1:1), 23.3 g (33 %) of 3a were obtained 3b was obtained from the filtrate of the crystallization of 3a, thus the filtrate was evaporated to dryness and the residue recrystallized from toluene/heptane (3:1) to afford 20.5 g (29.3 %).
3.2 Preparation of (-)-2-(4-chlorophenyl)-3-methylbutric acid (4)
The hydrolysis of 3a (21.25 g, 0.1 mol)with 450 ml of HCl (6 mol?dm-3) under reflux for 17 h led to 8.3 g (75 %) of pure 4 ([a]25D = -57.2, CHCl3, c=1.37, e.e.99 %) after recrystallization from petroleum ether.
3.3 Preparation of (S)-(-)-2-(4-chlorophenyl)-3-methylbutyl acyl chloride (5)
To thionyl chloride (6 ml, 0.04 mol) in dry toluene was added 4 (2.11 g, 0.01 mol) and the resulting solution was refluxed for 2 h. Excessive thionyl chloride was removed under reduced pressure; the left liquid must not be purified and can be used for the further reaction.
3.4 General procedures for the preparation of the target compounds 9a-9i were as follows
4,6-disubstituted-2-amino-pyrimidine (0.01 mol) was dissolved in 10 ml HCl (6 mol/l), potassium sulfocyanide (0.01 mol, 0.97 g) was added and the mixture was stirred at 90 ~ 100 ℃ for 6 h, then the reaction mixture was left to cool. The solid precipitation (7) was collected. Through the dropping funnel, (S)-(-)2-(4-chlorophenyl)-3-methylbutyl acyl chloride (5) was added to a solution of the (7) in 10 % NaOH. The mixture was allowed to 40 ~ 50 ℃ and stirred at this temperature until the reaction finished (controlled by TLC). The solution was then cooled in an ice-bath. The precipitate (8), which formed by cooling was collected. A mixture of compounds (8) and equimolar H2O2 in water was stirred at 0 ~ 2 ℃ using of TBAB catalyst to the mixture and the mixture continued to stirred at ambient for 1 h to ensure complete conversion to generate (9), recrystallized from DMF-EtOH-H2O to yield compound (9). Yield, m.p., elemental analysis, 1H NMR and Optical rotatios date of the compounds 9 are given in Table 2, 3 and 4. REFERENCES
[1] Koren, B.; Stanovnik, B.; Tisler, M.. J. Hterocyclic Chem. 1977, 14, 621~625
[2] Aoki, I.; Kuragano, T.; Okajima, N.; et al. EP 220695, 1987 (C.A.:107, P176057)
[3] Upadhyaya, J. S.; Sriuastava, P. K.. J. Indian Chem. Soc. 1982, 59 (6): 767
[4] Stetter, J..<ed.> In Herbicides Inhibiting Branched Chain Amino Acid Biosynthesis, Chemistry of Plant Protection 10, Springer, 1994
[5] Stidharm, M. A.. Weed Sci.1991, 39, 428.
[6] Schloss, J. V.; Ciskanik, L. M.; Dyk, D. V.. Nature, 1988, 331, 361.
[7] Abell, L. M.; Hanna, W. S.; Kunitaky, K. J.; et al. J. A. Pestic. Sci.1995, 44 (1): 89.
[8] Kamala, K. P.; Tayaprasad, R.; Reddy, K.. Synth. Commu. 1989, 13-14, 2621-2626.
[9] Elliott, M.; Farnham, A. W.; Janes, N. F.. Nature 1974, 248, 710
[10] Herve, J. J.; Motillon, J.. Fr. Demande, 2461458, 1981 (C.A.:1981, 96,2150e)
[11] Crammer, B.; Goldschmidt, Z.; Ikan, R.; et al. J. Agri. Food Chem. 1985, 33, 1148
[12] Griecw, P. A.. Organic synthesis in water, Blackie Acadeemic and professional, London, 1998
[13] Li, C. J.; Chang, T. H.. Inorganic Reactions In Aqueous Media, Wiley, New York, 1997
[14] Menicagli, R.; Malanga, C.. Synth. Commun.1994, 24 (15): 2153 水相中绿色合成(S)-(-)-5,7-二取代-2-(4-氯苯-3-甲基丁酰胺)-2H-1,2,4-噻二唑[2,3-α]-嘧啶衍生物
张斯勇 赵卿飞
(上海师范大学化学系,上海市,200234,中国)
摘要 以(S)-(-)-α-甲基苯甲胺为手性拆分剂,分离出纯光学活性(S)-(-)-2-(4-氯苯)-3-甲基丁酸,然后在水溶液中,以双氧水为氧化剂,四丁基溴化胺做为相转移催化剂,合成了一系列的新的(S)-(-)-5,7-二取代-2-(4-氯苯-3-甲基丁酰胺)-2H-1,2,4-噻二唑[2,3-α]嘧啶衍生物,通过红外,氢核磁,元素分析确定化合物结构。此方法与传统的方法比较具有反应时间短,产率高,环境友好等特点。
关键词 绿色合成,四丁基溴化胺,嘧啶衍生物