http://www.chemistrymag.org/cji/2002/044019pe.htm |
Apr. 1,
2002 Vol.4 No.4 P.19 Copyright |
Kinetics of direct vapor-phase carbonylation of methanol on sulfided Mo/C catalyst
Peng Feng(Department of Chemical Engineering, South China University of Technology, GuangZhou 510641, China)
Received July 30, 2001; Supported by National Natural Science Foundation of China (29903003)
Abstract Direct Vapor-phase
carbonylation of methanol was investigated under atmospheric pressure. A novel
heterogeneous sulfided Mo/C catalyst was used for the reaction without addition of CH3I
promoter to the feed. Experiments were designed with the elimination of mass-transfer
resistances to extraction data. The data of primary reactions in the carbonylation were
collected with a differential tubular reactor. Feed composition to the reactor was changed
to examine the effect of partial pressures of reactants on the reaction rate. Power law
rate models were employed to express the conversion of methanol and the yields of methyl
acetate and methane. Adequate results were obtained with the models to represent the
experimental data.
Keywords methanol carbonylation; vapor phase carbonylation;
kinetics; Mo/C catalyst; supported catalyst; methyl acetate.
Methanol carbonylation has attracted significant attention in C1 chemistry. Monsanto process for acetic acid via homogeneous carbonylation of methanol is an example of the largest scale commercial application of this route [1,2]. The significance of acetic acid in the petrochemical industry has drawn an extensive range of activities in industrial and academic sectors for a long-time. In order to improve the current technology, the use of a heterogeneous catalyst seemed more promising. Many research efforts have been indulged in the search for an appropriate catalyst to carry out vapor phase carbonylation under atmospheric pressure; for example, supported rhodium complexes, and Ni/C、Ni-Sn/C catalysts [3-6]. Nickel based catalyst was found to exhibit satisfactory activity and selectivity for the carbonylation of methanol. Methyl iodide was found to be essential in the cases; the rate-determining step of the reaction was discovered to be the cleavage of the C-I bond of methyl iodide. However, there has been very little success in finding heterogeneous or homogeneous catalysts that can operate effectively without using a halide promoter [7,8]. In our previous work direct carbonylation of methanol without any halide in the feed as promoter is reported. A novel Mo/C catalyst has been found to have a high activity and selectivity for the vapor phase carbonylation of methanol to methyl acetate without addition of CH3I promoter to the feed [9-11].
In this paper, kinetic analysis of a vapor-phase methanol carbonylation utilizing this novel catalyst is presented. Pore and external film resistances were eliminated in the reaction experiments. Power law models represent the primary reaction data. Parameters of the models were determined from the data by means of linear regression.
2. EXPERIMENTAL
2.1 Catalyst Preparation
The catalyst was prepared via incipient impregnation. Predetermined quantities of ammonium
heptamolybdate (AR grade) were dissolved in deionized water. A commercially available
granular activated carbon (olive based, in 20-40 mesh particle size,1000m2/g,
provided by Beijing Activated Carbon Factory) was then impregnated with the solution for
4-6h.The slurry was sulfidized in (NH4)2S aqueous solution (S≥8%
mass) for 4h. Then the catalyst precursor was dried in an air oven at 393K for 12h. Prior
to the catalytic tests, the dried catalysts were treated in situ with H2 at
673K for 2h. The Mo content in the catalyst was 10% (mass).
2.2 Carbonylation
Methanol carbonylation was carried out in a fixed bed reactor with a continuous flow
system at atmospheric pressure. The catalyst bed contained a mixture of catalyst (0.5g)
and powdered quartz (0.5g). The latter was employed to diminish both the gas channeling
and the temperature gradient in the bed. Each carbonylation experiment was initiated with
catalyst reduction performed in situ at 673 K under flowing H2 for 2 h. The
temperature of the catalyst bed was then decreased to the desired reaction temperature.
Reactants were then continuously fed at the temperature to activate the catalyst. Kinetic
data were not collected until the catalyst was activated. In a typical run, the data were
collected after the catalyst was on stream for 3 h. The partial pressures of methanol,
carbon monoxide in the carbonylation were changed via regulation of reactant flow rates.
Carbon monoxide and nitrogen were fed from gas cylinders. The flow rates of the gases were
set by means of mass-flow controllers. The independent partial pressure variation was
accomplished through usage of nitrogen in the feed, which compensated for variation of a
reactant. The conversion of methanol was limited to less than 8% so as to obtain a
differential mode of operation, which furnished the advantage of minimizing interference
caused by carbonylation products. The complicated data analysis due to concentration and
temperature gradients was also markedly simplified.
The effluent gas from the reactor was sampled by an auto-sampling valve
and was immediately analyzed in a gas chromatograph. Methanol conversion and yield for the
reaction are defined as the reference [9].
3.1 Stability of catalyst
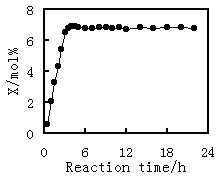
The conversion of methanol on novel catalyst steadily increases with the time on stream (Figure 1). The activation process lasts for about 3 h. the activity of the catalyst reaches a maximum at the end of the activation. No obvious deactivation is observed for the catalyst for at least 20 h after the activation has completed. Data for kinetic examinations are collected with the catalyst on stream during 3-20h. The data are, therefore, considered as steady-state data.
 |
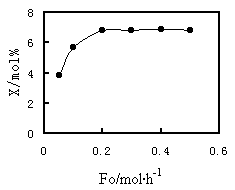 |
Fig.1 Stability of catalyst |
Fig.2 Effect of total molar flow rate on methanol conversion |
Reaction condition: T=573K; P=100kPa; contact time=0.85gcat·h/mol; CO/MeOH/N2=4/1/5 |
|
3.2 External film and pore resistance
The significance of external film resistance
was examined. The results are illustrated in Figure 2. The data in this figure were
obtained by passing reactants through the catalyst bed at varied rates of feed. The
reaction temperature and the contact time were maintained constant during obtaining of
these data. If the external film resistance is insignificant, the conversion is expected
to be independent of the flow rate. Otherwise, external film resistance is present.
External film resistance, as indicated in Figure 2, can be neglected at a rate of flow
exceeding 0.2 mol/h.
The relative significance of external film resistance and surface
reaction toward the overall rate of reaction can also be managed via reaction temperature.
Diminishing the reaction temperature decreases the rate of surface reaction more than the
rate of transport through the film. As a consequence, decreased reaction temperature leads
to a declined influence of external film and shifts the overall reaction toward surface
reaction control. The insignificance of external film resistance is illustrated in Figure
2 at a flow rate of 0.2 mol/h and at a reaction temperature of 573 K. The experiments for
kinetic data were therefore performed with a minimum flow rate of 0.2 mol/h and a maximum
reaction temperature 573 K. The enforcement of the conditions ensured the obtained data
were outside the influence of external film resistance.
Pore resistance produces both concentration and temperature gradients
within a catalyst that distort the rate data to yield erroneous kinetic conclusions. The
utilization of a small catalyst can diminish the concentration and temperature variations
between the center and surface of the catalyst. The experiment shows that pore resistance
can be neglected at the particle of catalyst less than 40 mesh.
3.3 Rate model
Many reactions occur during carbonylation of methanol. The
reactions form a complicated reaction path network involving several parallel and
consecutive routes. The reactions may be reversible or irreversible, and both reactants
and products can participate in various stages. The complicated situation is significantly
simplified by means of a differential reactor, which enables collection of kinetic data
with a small conversion of methanol. Therefore, reversible reactions and reactions
involving products can be neglected; only primary reactions need be considered in these
conditions. Selectivity of dimethyl ether is lower as 1% in this catalytic system;
therefore the kinetics of formation of dimethyl ether is not discussed.
The rate of conversion of methanol and the rates of productions of
methyl acetate and methane were represented by power law models of the following form:
r=kPca Pmb
Where r = reaction rate (mmol/(h· g-cat)), k = rate constant, Pc =
partial pressure of carbon monoxide (kPa), Pm = partial pressure of methanol
(kPa), and a, b = reaction orders.
Furthermore, the rate constant is expressed in terms of reaction
temperature according to Arrhenius relation: k=koexp (-E/RT).
Where k0 = frequency factor, E = activation energy, R = gas
constant, and T = reaction temperature (K).
The resulting equation can be transformed into: ln r=ln ko-
E/RT + alnPc + blnPm .
The parameters, k0, E, a and b were determined from the
method by means of linear regression.
3.4 Effect of partial pressures
The rates of ethanol conversion and product
formation on the different partial pressures of reactants at 543 K were plotted in Figures
3 and 4. Increasing carbon monoxide partial pressures increases the rates of methanol
conversion and methyl acetate formation; especially the formation rate of methyl acetate
is greatly affected by the partial pressure of carbon monoxide. However, the formation
rate of methane decreases markedly with increasing carbon monoxide partial pressures. On
the other hand, the formation rates of both methyl acetate and methane increase markedly
with increasing the partial pressure of methanol. And then the effects of methanol partial
pressures on the rates of methanol conversion and product formations are nearly the same.
The same results are found at the reaction temperature of 513 K and 573 K. The reaction
orders and rate constants were determined by means of linear regression. The parameters
determined from the regression are listed in Table 1.
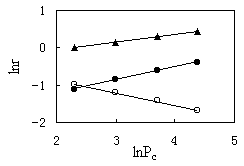 |
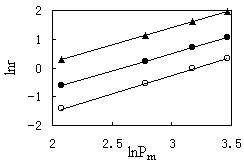 |
Fig.3
Effect of partial pressure of CO at 543 K |
Fig.4 Effect
of partial pressure of methanol at 543 K |
|
|
Table 1 Reaction orders and rate constants of methanol conversion and product formations
Rates |
rate constants,k |
Reaction orders |
|||
513k |
543k |
573k |
a |
b |
|
MeOH conversion |
10.3*10-3 |
5.33*10-2 |
2.37*10-1 |
0.20 |
1.21 |
CH4 formation |
5.35*10-3 |
5.59*10-2 |
5.20*10-1 |
-0.34 |
1.30 |
| MeCOOMe formation | 3.21*10-3 |
1.25*10-2 |
0.43*10-1 |
0.35 |
1.19 |
As shown in table 1, the
reaction orders of methanol conversion and product formation with respect to methanol
exceed 1.0, showing the novel catalyst depended heavily on the methanol. The entirely
different results were observed for methanol carbonylation with addition of methyl iodide
promoter to the feed [5]. The production of methyl acetate has the smallest
reaction order with respect to methanol and the largest reaction order with respect to
carbon monoxide; hence decreased partial pressure of methanol leads to increasing
selectivity of methyl acetate. The selectivity of methyl acetate can be further increased
with an increased partial pressure of CO and a decreased reaction temperature.
According to the reaction orders, the reaction mechanism of direct
vapor-phase carbonylation of methanol is analyzed. The initial step of this reaction is
suggested to be the cleavage of methanol to form CH3+, followed by
adsorbed CO insertion into the metal-methyl bond, forming CH3C (O)+,
which further interacts with methanol to form methyl acetate [12]. The marked
difference between direct and indirect carbonylation is the reaction order with respect to
the partial pressure of methanol. Methyl iodide was found to be essential in the former,
the reaction order of the promoter ( CH3I) is 1-1.5 and that of methanol is
0-0.6. The reaction orders observed with respect to methyl iodide partial pressure on a
series of catalysts are mostly first order, hence the rate-determining step of the
reaction is suggested to be the cleavage of the C-I bond of methyl iodide [3,5].
However, in the absence of methyl iodide, the reaction rates of direct carbonylation
depend strongly on the partial pressure of methanol. It is reasonable to assume that the
rate-determining step of the reaction is the cleavage of methanol.
3.5 Activation energy of the reactions
Activation energy of the reactions were determined from the
rate constants data obtained at different reaction temperatures. Arrhenius plot of
reaction rates for methanol carbonylation was shown in Figure 5. The rate constants of
methanol conversion and the product formation exactly corresponded with Arrhenius
equation. The parameters (k0 and E) determined from the Figure 5 are listed in
Table 2. With the halide in the feed as promoter, homogeneous and heterogeneous catalysts
were both utilized for the carbonylation of methanol [5]. The reaction
activation energies appeared to be dependent of the catalyst type and the reaction
conditions, activation energy of the methanol reaction was reported from 37.4KJ·mol-1
to 90.4KJ·mol-1. While without any halide in the feed as promoter, the
activation energy of methanol carbonylation reached 128 KJ·mol-1. The result
demonstrated that the direct carbonylation reaction was more difficult to occur than the
indirect carbonylation.
 |
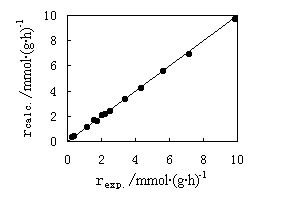 |
| Fig.5 Arrhenius plot of reaction rates for methanol carbonylation | Fig.6 Comparison of methyl acetate formation rate sbetween experimental and calculated valves |
|
|
Table 2
The data of k0 and E for methanol carbonylation reactionRates |
ko |
E /kJ·mol-1 |
MeOH conversion |
1.1*1011 |
128 |
CH4 formation |
5.8*1016 |
187 |
MeCOOMe formation |
2.0*108 |
106 |
The adequacy of the models was examined by plotting predicted formation rates of methyl acetate according to the model versus experimental rates (Fig. 6). The points exactly lie on the diagonal line for a completely precise model. The relative error of calculated results and experimental ones for methyl acetate formation rate is within 6.5%. The models developed in this investigation are represented in accord with the experimental data.
4 CONCLUSIONS
Power law models were developed for primary
reactions of carbonylation of methanol on a novel sulfided Mo/C catalyst. The kinetic
parameters for the direct vapor phase carbonylation of methanol were different from those
of the indirect carbonylation of methanol with the promoter of methyl iodide. The reaction
orders of methanol conversion and the product formation with respect to methanol exceed
1.0, showing the novel catalyst depended heavily on methanol. The production of methyl
acetate has the smallest reaction order with respect to methanol and the largest reaction
order with respect to carbon monoxide. The reaction orders of methyl acetate formation
with respect to methanol and CO were respectively 1.19 and 0.35; and the reaction
activation energy was 106 kJ/mol. Sufficient accuracy was obtained with the model to
determine the formation rate of methyl acetate.
[1] Roth J F, Craddock J H. Chem Tech, 1971, 23: 600.
[2] Lowry R P, Aquilo A. Hydrocarbon Proce, 1974, 53 (11): 106.
[3] Omata K, Fujimoto K, Shikada T, Tominaga H. Ind Eng Chem Prod Res Dev, 1985, 24 (2): 234.
[4] Liu T C, Chiu S. Appl Catal, 1994, 117 (1): 17.
[5] Liu T C, Chiu S. Ind Eng Chem Res, 1994, 33: 1674.
[6] Takahashi N, Orikasa Y, Yashima T, etal. J Catal, 1979, 59 (1): 61.
[7] Wegman R W. J Chem Soc Chem Commun, 1994, (8): 947.
[8] Calafat A, Laine J. Catal Lett, 1994, 28 (1): 69.
[9] Peng F. Chem J Internet, 2000, 2 (7): 32.
[10] Peng F, Zheng G S. Chin J Fuel Chem Technol (Ranliao huaxue xuebao), 2000, 28 (4): 340.
[11] Peng F, Li X H, Li J W. Chin J Fuel Chem Technol (Ranliao huaxue xuebao), 1999, 27 (6): 562.
[12] Peng F, Ren Y Q. Chin Natural Gas Chem Ind( Tianranqi huagong), 2001, 26 (3): 15.