http://www.chemistrymag.org/cji/2002/045021pc.htm |
Apr. 1,
2002 Vol.4 No.5 P.21
Copyright |
Synthesis of Poly(ethyl acrylate)-methanofullerene[60] via the macromolecular phosphonium ylid
Jin Hongmei, Shen
Weiping , Zhu Yun
(Department of Chemistry, College of science, Shanghai
University, Shanghai 200436,China)
Abstract A new
preparation of the Poly(ethyl acrylate)–methanofullerene[60]
and the multi-branched Poly(ethyl acrylate)-methanofullerene[60] is developed by using the
Poly(ethyl acrylate)triphenylphosphonium chloride and the star shape of Poly(ethyl
acrylate)triphenylphosphonium chloride via the macromolecular phosphonium ylid. They are
characterized by size-exclusion chromatography , UV-VIS. and 1H-NMR spectra.
Keywords Poly(ethyl acrylate)triphenylphosphonium chloride ;
macromolecular phosphonium ylid ; Poly(ethyl acrylate)-methanofullerene[60]
(上海大学理学院化学系 上海 200436)
2002
年1月10日收稿 国家自然科学基金资助项目(No.291000021)摘要
: 用氯化三苯基鏻聚丙烯酸乙酯和星形氯化三苯基鏻聚丙烯酸乙酯,经过大分子膦Ylid制备新的富勒烯[60]聚丙烯酸乙酯衍生物和含富勒烯[60]的多分支状聚丙烯酸乙酯衍生物。并对它们进行了GPC、UV-VIS、1H-NMR光谱鉴定。关键词:(星形)氯化三苯基鏻聚丙烯酸乙酯;大分子膦Ylid;富勒烯[60]聚丙烯酸乙酯衍生物
富勒烯衍生物特别是高分子化衍生物,因其结合了富勒烯[60]优良的光、电、磁性能和高分子化合物的成材性及协同效应,所以在材料科学特别是微电子、信息材料方面它将起着不可估量的作用[1]。富勒烯[60]亚甲基(methanofullerene)衍生物是合成高分子化富勒烯[60]的一种主要研究手段。它主要利用富勒烯[60]球烯上的[6,6]双键与带有良好离去基团的碳负离子发生加成-消除反应,结果得到富勒烯[60]衍生物[2]。作者曾报道了用基团转移聚合方法制备含氯化三苯基鏻端基的聚丙烯酸酯功能高分子[3]。本文所述用此大分子鏻盐经过膦Ylid反应合成富勒烯[60]聚丙烯酸酯衍生物。
1 实验部分1.1 实验材料
溶剂二氯甲烷、石油醚、甲苯、四氢呋喃(THF)都经过严格的无水处理。试剂三苯膦真空加热到700C干燥12小时,然后通入氩气密封保存。三甲基氯硅烷(TMCS)需用时加入氢化钙,在氩气保护下常压蒸馏,收集570C馏份.再放入一些氢化钙密封保存。溴化锌真空强热(2000C)干燥8小时,然后在氩气氛下,真空升华得溴化锌白色细晶体,密封保存。单体丙烯酸乙酯(EA)用10%的氢氧化钾液洗涤2~3次,以除去阻聚剂.然后加入无水硫酸镁干燥过夜.过滤后用氢化钙干燥过夜.过滤后在氩气保护常压蒸馏收集990C中心馏份,加入氢化钙密封保存。交联剂甲基丙烯酸二乙二醇酯用5%的氢氧化钾溶液洗涤,用无水硫酸镁干燥,过滤后用氢化钙干燥,使用前再经中性氧化铝层析。富勒烯[60]购于武汉大学,纯度99.99%。二异丙胺锂(LDA)购于ACROS,2M THF溶液。
1.2 星形氯化三苯基鏻聚丙烯酸乙酯的制备
将溴化锌(1.5g,6.66mmol)(催化剂约为单体的16~18mol%为好)放入100ml三颈瓶中。真空强热下(注意防止溴化锌熔化),干燥半小时,以除去痕量水份。通入氩气冷却到室温。在氩气保护下,注射入丙烯酸乙酯(3.8ml,35.07mmol)和10ml二氯甲烷,磁力搅拌下溶解溴化锌。当溶液由浑浊变澄清时,用冰盐浴冷却至-50C.将已配好的三苯膦(0.5g,1.91mmol)和三甲基氯硅烷(0.25ml,1.97mmol)作为引发剂的二氯甲烷(5ml)溶液用针筒注入反应体系中.约过1~3分钟,体系温度自然上升至300C左右。体系粘度增加并呈粉红色(可加入适量二氯甲烷,以利于搅拌)。约10分钟以后,温度降到00C,继续搅拌一小时。然后,注入甲基丙烯酸二乙二醇酯交联剂(0.5ml,2.65mmol),体系温度略上升0.5~l0C,随后,在00C下继续搅拌,反应5小时后加入甲醇终止反应。真空除去部分二氯甲烷.然后加入大量石油醚,沉淀析出聚合物。倾去清液,再加入大量无水乙醚,静置过夜。然后过滤蒸去溶剂,得4.52克粘稠状淡黄色聚合物。得率接近100%。GPC;Mn=14083(D=1.19)。1H-NMR(CDCl3);7.76~7.71(m,ArH);4.13(q,J=6.5Hz -COOCH2CH3); 2.18~1.51(m,-CH2CH-);1.23ppm (t,J=6.5Hz -COOCH2CH3)。氯化星形三苯基鏻聚丙烯酸乙酯结构表征工作另见报道。
1.3 富勒烯[60]聚丙烯酸乙酯衍生物的合成
将2.26g氯化三苯基鏻聚丙烯酸乙酯(GPC Mn:3368,每克聚合物含氯化三苯基鏻端基0.48mmol,总1.08mmol)溶于35mlTHF中,置于250ml三颈瓶中,通氩气保护,磁力搅拌,冷却至-78oC(丙酮-干冰浴)随后,用注射器注入0.58ml LDA(2M/THF溶液)。 反应40分钟后,滴入溶有130毫克富勒烯[60](0.18 mmol)的甲苯溶液150毫升。干冰慢慢挥发掉,温度升至室温,继续搅拌反应48小时。将反应液过滤后,减压蒸去部分溶剂至剩余约10ml残余液时,将其加入到大量石油醚中沉淀出聚合物,再用二氯甲烷溶解,过滤而后用石油醚沉出聚合物。干燥后得1.76克(GPC;Mn=9066)。
同样用2.5克星形氯化三苯基鏻聚丙烯酸乙酯(GPC;Mn=14083,每克聚合物含氯化三苯基鏻端基0.42mmol,总1.06mmol ) 重复上述合成过程。最后得到深棕色粘稠胶状物2.13克(GPC;Mn=88945)。
2 结果与讨论
2.1 (星形)氯化三苯基鏻聚丙烯酸乙酯和富勒烯[60]的反应
Scheme 1
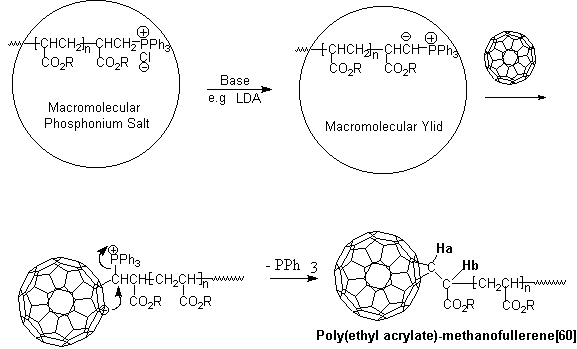
富勒烯[60]是一个缺电子烯烃。它能接受1~6个电子,易被亲核试剂进攻[4]。Scheme1显示了(星形)氯化三苯基鏻聚丙烯酸乙酯----大分子鏻盐,在强碱如二异丙胺锂(LDA)的作用下,被夺取质子形成大分子膦Ylid。随后对富勒烯[60]进行亲核进攻生成一个新的碳负离子(中心碳原子在富勒烯[60]球壳上)。新的碳负离子紧接着发生亲核取代反应,消除三苯膦生成富勒烯[60]亚甲基衍生物即富勒烯[60]聚丙烯酸乙酯衍生物。由于富勒烯[60]球面上存在着多个“反应点”能接受六个电子形成六价C60负离子和形成多取代富勒烯[60]衍生物[5],因此设计大分子鏻盐与富勒烯[60]反应的摩尔比为6∶1,使其能最大限度地加成到富勒烯[60]球面上去,期望在富勒烯[60]球面上形成六个接枝聚丙烯酸乙酯的高分子链。运用星形大分子鏻盐和富勒烯[60]反应时,鏻盐与富勒烯[60]反应的摩尔比也过量。希望利用富勒烯[60]球面上的多点反应性能形成一个交联核心。从而制备含富勒烯[60]的高度分支状聚丙烯酸乙酯衍生物(Scheme2)。
Scheme 2
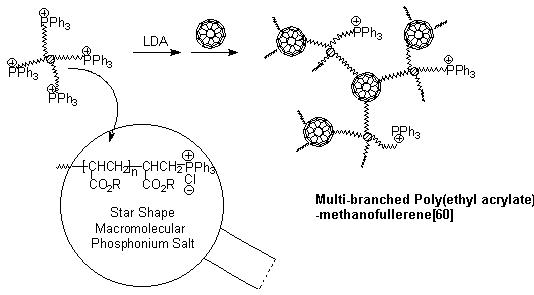
在反应中,用TLC方法跟踪反应发现确实有三苯膦生成。得到的富勒烯[60]聚丙烯酸酯衍生物是个深棕色的粘稠油状物,溶于二氯甲烷、丙酮、氯仿等中等极性溶剂。而未反应的富勒烯[60]不溶于这些溶剂,所以在产物的纯化过程中未反应的富勒烯[60]很容易被分去。但未反应的氯化三苯基鏻聚丙烯酸乙酯却很难从产物中分离。
富勒烯[60]聚丙烯酸酯衍生物为深棕色透明的粘稠油状物,溶于丙酮后加入硅烷偶联剂可制成膜。
2.2 富勒烯[60] 聚丙烯酸酯衍生物的结构表征
2.2.1 GPC 测定
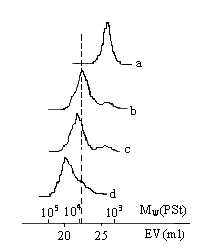 |
Fig.1
GPC of the Polymers |
用THF作溶剂对氯化三苯基鏻聚丙烯酸乙酯(a);富勒烯[60]聚丙烯酸乙酯衍生物(b);星形氯化三苯基鏻聚丙烯酸乙酯(c);富勒烯[60]多分支状聚丙烯酸乙酯衍生物(d)等四个聚合物做了GPC测定(Fig.1),分析了它们的相对分子量,发现聚合物a经膦Ylid反应以后得到聚合物b,它们的分子量也相应地从3368增加到9066。这说明在富勒烯[60]的球面上大约接上了3个聚丙烯酸乙酯的链。同时从GPC的峰形上可看出尚有未反应的聚合物a存在。同样用星型聚合物c进行反应,它们的分子量从原有的星型聚合物的14000左右改变为接近90000。这反映了我们得到了所预期的把富勒烯[60]作为交联核心的多分支状聚丙烯酸乙酯衍生物。
2.2.2 UV-Visible光谱测定
 |
Fig.2
UV-Visible of the Polymers |
含富勒烯[60]的多分支状聚丙烯酸乙酯衍生物(b)、星形氯化三苯基鏻聚丙烯酸乙酯(c)及富勒烯[60](a)的二氯甲烷溶液的紫外光谱(Fig.2)显示聚合物星形氯化三苯基鏻聚丙烯酸乙酯(c)在227nm处有一明显吸收,并且在240-290nm处有一个宽的弱吸收峰,而富勒烯[60](a)在262nm、290nm和330nm处有吸收峰。富勒烯[60]的星形聚丙烯酸酯衍生物(b)的吸收发生在238nm、260nm以及328nm处,其中238和260nm的峰强度明显增强。由此猜想可能是由于球壳烯与聚合物链之间的π-π相互作用增强的结果。这也说明了星形聚丙烯酸酯成功地接枝到了富勒烯[60]的球面上,同时在这个大分子中还存在未和富勒烯[60]反应的氯化三苯基鏻端基。
2.2.3 1H-NMR谱测定
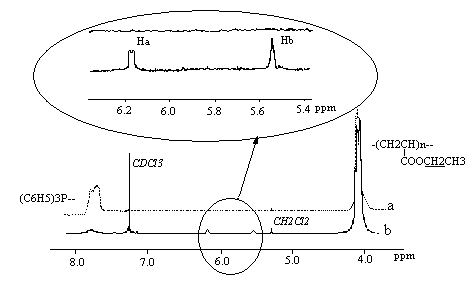
Fig.3 1H-NMR of the polymers
a) Poly(ethylacrylate)triphenylphosphonium chloride
b) Poly(ethylacrylate)-methanofullerene[60]
得到的富勒烯[60]聚丙烯酸酯衍生物是个深棕色的粘稠油状物,溶于氘代氯仿后,用1H
NMR 光谱鉴定(Fig.3)。发现与氯化三苯基鏻聚丙烯酸乙酯的1H
NMR
光谱比较,原来三苯基鏻端基的芳质子吸收强度明显减弱与聚丙烯酸乙酯链质子的吸收强度之比减小了许多[6]。这反映了氯化三苯基鏻聚丙烯酸乙酯这一大分子鏻盐在LDA的作用下形成大分子膦叶立德,然后对富勒烯[60]进行亲核进攻并脱去三苯膦小分子。而未反应的大分子鏻盐在1H
NMR 光谱中出现残存的吸收。同样和反应物相比在6.17ppm和5.54ppm处出现了两个新的弱吸收峰,经分析初步认定为富勒烯[60]球面上亚甲基和与之相连的次甲基上的质子Ha、Hb的吸收(见Scheme1)。这很好地证明了已成功合成富勒烯[60]聚丙烯酸乙酯衍生物[7]。另外,根据核磁谱上芳质子吸收与侧链酯基上亚甲基质子吸收强度之比的变化和GPC所测得的分子量增大的两倍多佐证,可粗略估计富勒烯[60]球面上平均接枝了三个聚丙烯酸乙酯支链。用星形大分子鏻盐反应所得的含富勒烯[60]多分支状聚丙烯酸乙酯衍生物其呈现的1H
NMR
光谱吸收与上述的线形大分子鏻盐作为反应物时基本一致。同样结合GPC所测相对分子量的变化,从原有的14000改变成90000左右。此证实了富勒烯[60]多分支状聚丙烯酸乙酯衍生物的制备成功。这些为我们今后研究这类既含有平面芳香环又拥有球形芳性的多分枝状聚合物的功能性提供了很好的材料[8]。
[1] Wang Y. Nature, 1992, 356: 585.
[2] Geckeler K E. Hirsch A. J.Am.Chem.Soc., 1993, 115: 3850.
[3] Shen Weiping. Jin Hongmei. Makromol.Chem., 1992, 193: 743.
[4] Hirsh A. Synthesis, 1995, 895.
[5] Wu M. Wei X. Qi L et al. Tetrahedron Lett., 1996, 37: 7409.
[6] Shi Z J, Shen W P, Yao Y et al. Chinese Journal of Magnetic resonance (Bopuxue Zazhi), 1992, 9 : 369.
[7] Bestmann H J, Hadawi D et al. Tetrahedron Lett., 1994, 35: 9017.
[8] Cloutet E, Gnanou Y et al. Chem.Commun., 1996, 1565.